cAMP Pathway
G-Protein Coupled Receptors
G protein-coupled receptors (GPCRs), also known as seven transmembrane domain receptors, 7TM receptors, heptahelical receptors, serpentine receptor, and G protein-linked receptors (GPLR), comprise a large protein family of transmembrane receptors that sense molecules outside the cell and activate inside signal transduction pathways and, ultimately, cellular responses. G protein-coupled receptors are found only in eukaryotes, including yeast, plants, choanoflagellates, and animals. The ligands that bind and activate these receptors include light-sensitive compounds, odors, pheromones, hormones, and neurotransmitters, and vary in size from small molecules to peptides to large proteins. G protein-coupled receptors are involved in many diseases, and are also the target of around half of all modern medicinal drugs. G-Protein coupled receptors called so because its ability to bind with trimeric G-proteins (Guanosine nucleotide binding proteins).
Structure of GPCRs:
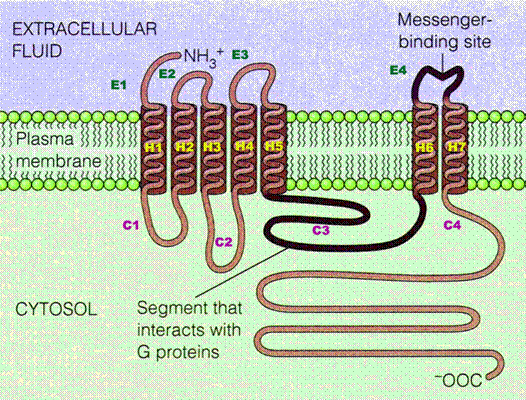
GPCRs are integral membrane proteins that possess seven membrane-spanning domains or transmembrane helices. The extracellular parts of the receptor can be glycosylated. These extracellular loops also contain two highly-conserved cysteine residues that form disulfide bonds to stabilize the receptor structure. In 2007, the first structure of a human GPCR was solved. All receptors of this type have the same orientation in the membrane and contain seven transmembrane -helical regions (H1–H7), four extracellular segments (E1–E4), and four cytosolic segments (C1–C4). The carboxyl-terminal segment (C4), the C3-loop and in some receptors, also the C2 loop are involved in interactions with a coupled trimeric G protein. G protein-coupled receptors are activated by an external signal in the form of a ligand or other signal mediator. This creates a conformational change in the receptor, causing activation of a G protein. Further effect depends on the type of G protein.
Ligands of GPCRs:
GPCRs include receptors for sensory signal mediators (e.g., light and olfactory stimulatory molecules); adenosine, bombesin, bradykinin, endothelin, γ-aminobutyric acid (GABA), hepatocyte growth factor, melanocortins, neuropeptide Y, opioid peptides, opsins, somatostatin, tachykinins, vasoactive intestinal polypeptide family, and vasopressin; biogenic amines (e.g., dopamine, epinephrine, norepinephrine, histamine, glutamate (metabotropic effect), glucagon, acetylcholine (muscarinic effect), and serotonin); chemokines; lipid mediators of inflammation (e.g., prostaglandins, prostanoids, platelet-activating factor, and leukotrienes); and peptide hormones (e.g., calcitonin, C5a anaphylatoxin, follicle-stimulating hormone (FSH), gonadotropic-releasing hormone (GnRH), neurokinin, thyrotropin-releasing hormone (TRH), and oxytocin). GPCRs that act as receptors for stimuli that have not yet been identified are known as orphan receptors. Whereas, in other types of receptors that have been studied, ligands bind externally to the membrane, the ligands of GPCRs typically bind within the transmembrane domain.
Physiological Roles of GPCRs:
GPCRs are involved in a wide variety of physiological processes. Some examples of their physiological roles include:
- The visual sense: the opsins use a photoisomerization reaction to translate electromagnetic radiation into cellular signals. Rhodopsin, for example, uses the conversion of 11-cis-retinal to all-trans-retinal for this purpose
- The sense of smell: receptors of the olfactory epithelium bind odorants (olfactory receptors) and pheromones (vomeronasal receptors)
- Behavioral and mood regulation: receptors in the mammalian brain bind several different neurotransmitters, including serotonin, dopamine, GABA, and glutamate
- Regulation of immune system activity and inflammation: chemokine receptors bind ligands that mediate intercellular communication between cells of the immune system; receptors such as histamine receptors bind inflammatory mediators and engage target cell types in the inflammatory response
- Autonomic nervous system transmission: both the sympathetic and parasympathetic nervous systems are regulated by GPCR pathways, responsible for control of many automatic functions of the body such as blood pressure, heart rate, and digestive processes
- Cell density sensing: A novel GPCR role in regulating cell density sensing.
Classification of GPCRs:
GPCRs can be grouped into 6 classes based on sequence homology and functional similarity:
! Class A (or 1) (Rhodopsin-like)
! Class B (or 2) (Secretin receptor family)
! Class C (or 3) (Metabotropic glutamate/pheromone)
! Class D (or 4) (Fungal mating pheromone receptors)
! Class E (or 5) (Cyclic AMP receptors)
! Class F (or 6) (Frizzled/Smoothened)
The very large rhodopsin A group has been further subdivided into 19 subgroups (A1-A19). More recently, an alternative classification system called GRAFS (Glutamate, Rhodopsin, Adhesion, Frizzled/Taste2, Secretin) has been proposed. The human genome encodes roughly 350 G protein-coupled receptors, which detect hormones, growth factors and other endogenous ligands. Approximately 150 of the GPCRs found in the human genome have unknown functions.
Regulation of GPCRs:
GPCRs become desensitized when exposed to their ligand for a prolonged period of time. There are two recognized forms of desensitization: 1) homologous desensitization, in which the activated GPCR is downregulated; and 2) heterologous desensitization, wherein the activated GPCR causes downregulation of a different GPCR. The key reaction of this downregulation is the phosphorylation of the intracellular (or cytoplasmic) receptor domain by protein kinases.
Phosphorylation by cAMP-dependent protein kinases
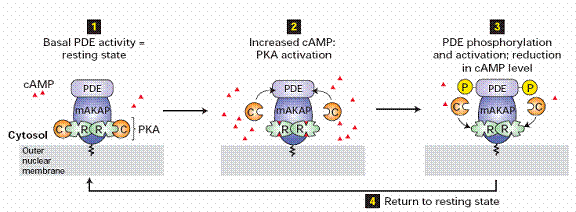
Cyclic AMP-dependent protein kinases (protein kinase A) are activated by the signal chain coming from the G protein (that was activated by the receptor) via adenylate cyclase and cyclic AMP (cAMP). In a feedback mechanism, these activated kinases phosphorylate the receptor. The longer the receptor remains active, the more kinases are activated, the more receptors are phosphorylated. In β2-adrenoceptors, this phosphorylation results in the switching of the coupling from the Go class of G-protein to the Gi class. cAMP-dependent PKA mediated phosphorylation is also known as heterologous desensitisation, because it is not specific to ligand bound receptor. In fact any receptor causing an increase in PKA activity will cause increased amounts of this type of desensitisation of other receptors coupled to Go (e.g., dopamine receptor D2 activation may lead to β2-adrenoceptor desensitisation of this type).
Phosphorylation by GRKs
The G protein-coupled receptor kinases (GRKs) are protein kinases that phosphorylate only active GPCRs. Phosphorylation of the receptor can have two consequences:
Translocation: The receptor is, along with the part of the membrane it is embedded in, brought to the inside of the cell, where it is dephosphorylated within the acidic vesicular environment and then brought back. This mechanism is used to regulate long-term exposure, for example, to a hormone, by allowing resensitisation to follow desensitisation. Alternatively, the receptor may undergo lysozomal degradation, or remain internalised, where it is thought to participate in the initiation of signalling events, the nature of which depend on the internalised vesicle's subcellular localisation.
Arrestin linking: The phosphorylated receptor can be linked to arrestin molecules that prevent it from binding (and activating) G proteins, effectively switching it off for a short period of time. This mechanism is used, for example, with rhodopsin in retina cells to compensate for exposure to bright light. In many cases, arrestin binding to the receptor is a prerequisite for translocation. For example, beta-arrestin bound to β2-adrenoreceptors acts as an adaptor for binding with clathrin, and with the beta-subunit of AP2 (clathrin adaptor molecules); thus the arrestin here acts as a scaffold assembling the componenets needed for clathrin-mediated endocytosis of β2-adrenoreceptors.
G-Proteins:
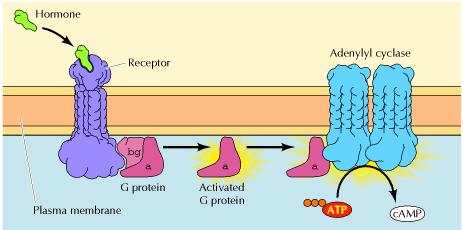
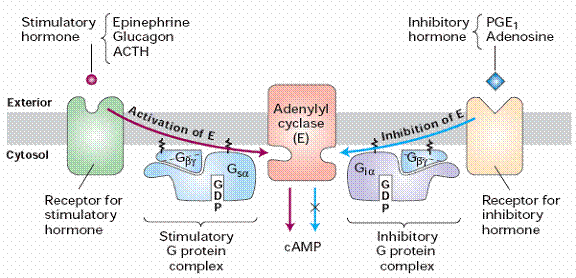
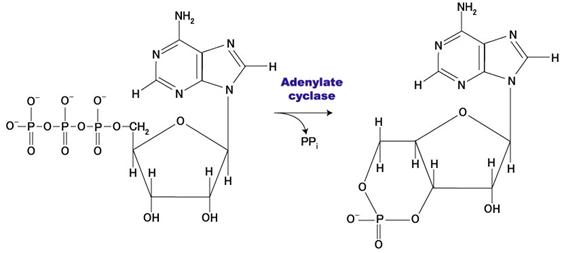
G proteins, short for guanine nucleotide-binding proteins, are a family of proteins involved in second messenger cascades. G proteins are so called because they function as "molecular switches," alternating between an inactive guanosine diphosphate (GDP) and active guanosine triphosphate (GTP) bound state, ultimately going on to regulate downstream cell processes. G proteins were discovered when Alfred G. Gilman and Martin Rodbell tried to figure out how adrenaline stimulated cells. They found that when a hormone like adrenaline bound to a receptor, the receptor did not stimulate enzymes like adenylate cyclase directly. Instead, the receptor stimulated a G protein, which then stimulated the adenylate cyclase to produce a second messenger, cyclic AMP. For this discovery they won the 1994 Nobel Prize in Physiology or Medicine. G proteins belong to the larger group of enzymes called GTPases.
Types of G-Proteins:
G protein can refer to two distinct families of proteins. Heterotrimeric G proteins, sometimes referred to as the "large" G proteins that are activated by G protein-coupled receptors and made up of alpha (α), beta (β), and gamma (γ) subunits. There are also "small" G proteins (20-25kDa) that belong to the Ras superfamily of small GTPases. These proteins are homologous to the alpha (α) subunit found in heterotrimers, and are in fact monomeric. However, they also bind GTP and GDP and are involved in signal transduction.
1. Heterotrimeric G-Proteins:
Heterotrimeric G proteins share a common mode of action, i.e., activation in response to a conformation change in the G-protein-coupled receptor, exchange of GTP for GDP and dissociation in order to activate further proteins in the signal transduction pathway. However, the specific mechanism differs between different types of G proteins. Receptor-activated G proteins are bound to the inside surface of the cell membrane. They consist of the Gα and the tightly associated Gβγ subunits. At the present time, four main families exist for Gα subunits: Gαs, Gαi, Gαq/11, and Gα12/13. These groups differ primarily in effector recognition, but share a similar mechanism of activation.

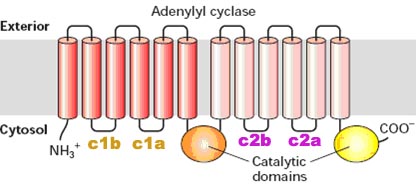
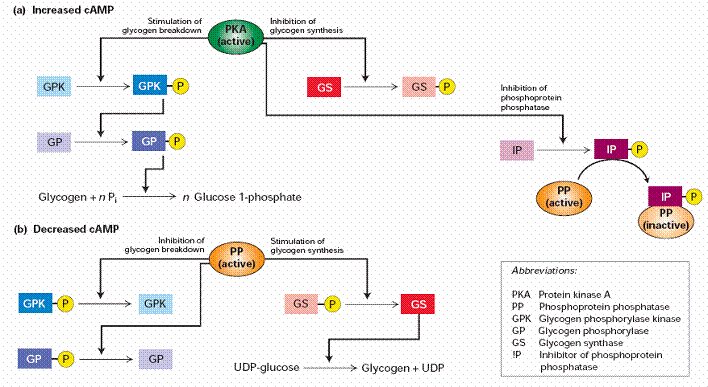
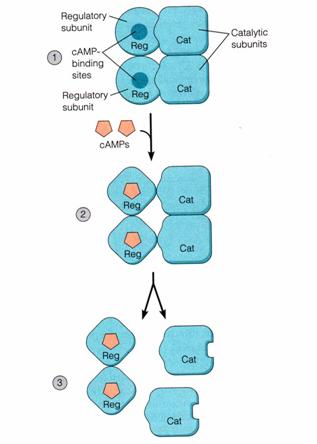
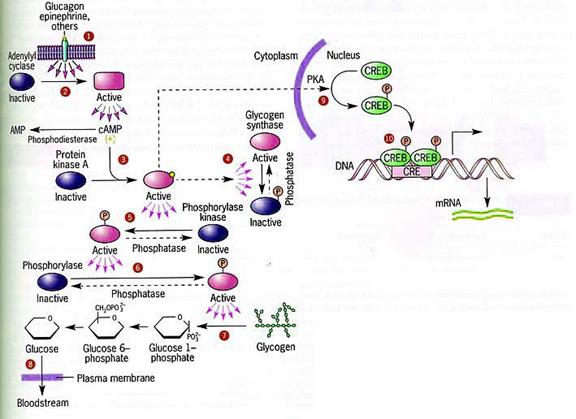
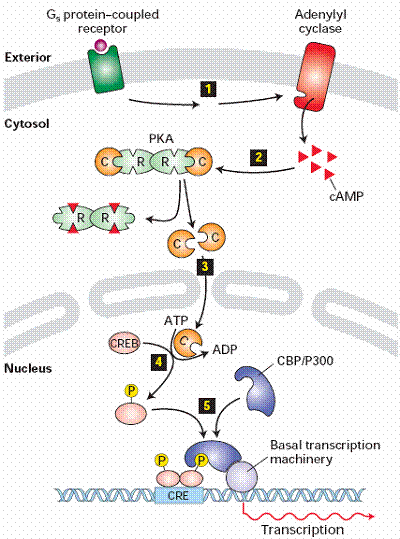
! Gαs stimulates the production of cAMP from ATP. This is accomplished by direct stimulation of the membrane-associated enzyme adenylate cyclase. cAMP acts as a second messenger that goes on to interact with and activate protein kinase A (PKA). PKA can then phosphorylate a myriad of downstream targets.
! Gαi inhibits the production of cAMP from ATP.
! Gαq/11 stimulates membrane-bound phospholipase C beta, which then cleaves PIP2 (a minor membrane phosphoinositol) into two second messengers, IP3 and diacylglycerol (DAG).
! Gα12/13 are involved in Rho family GTPase signaling (through RhoGEF superfamily) and control cell cytoskeleton remodeling, thus regulating cell migration.
! Gβγ sometimes also have active functions, e.g., coupling to L-type calcium channels.
2. Small G-Proteins (Small GTPase):
Small GTPases also bind GTP and GDP and are involved in signal transduction. These proteins are homologous to the alpha (α) subunit found in heterotrimers, but exist as monomers. They are small (20-kDa to 25-kDa) proteins that bind to guanosine triphosphate (GTP). This family of proteins is homologous to Ras GTPases and is also called the Ras superfamily GTPases.
Function of G-Proteins (G-Protein cycle):
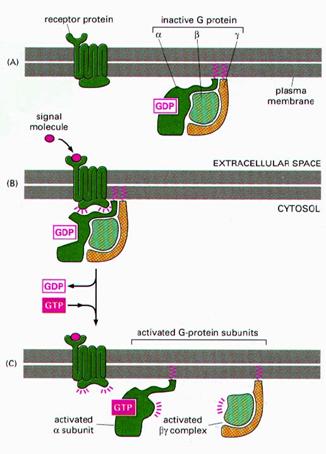
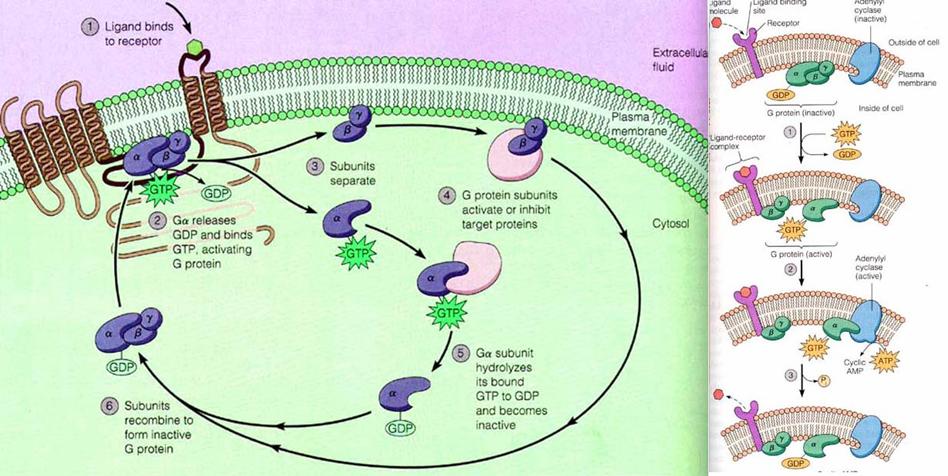
When a ligand activates the G protein-coupled receptor, it induces a conformation change in the receptor (a change in shape) that allows the receptor to function as a guanine nucleotide exchange factor (GEF) that exchanges GTP in place of GDP on the Gα subunit. In the traditional view of heterotrimeric protein activation, this exchange triggers the dissociation of the Gα subunit, bound to GTP, from the Gβγ dimer and the receptor. Both Gα-GTP and Gβγ can then activate different signaling cascades (or second messenger pathways) and effector proteins, while the receptor is able to activate the next G protein.
Once the exchange of nucleotides has occurred, the Ga·GTP complex dissociates from the Gbg subunit, but both remain anchored in the membrane. In most cases, Ga·GTP then interacts with and activates an associated effector protein. This activation is shortlived, however, because GTP bound to G is hydrolyzed to GDP in seconds, catalyzed by a GTPase enzyme that is an intrinsic part of the G subunit. The resulting Ga·GDP quickly reassociates with Gbg , thus terminating effector activation. In many cases, a protein termed RGS (regulator of G protein signaling) accelerates GTP hydrolysis by the G subunit, reducing the time during which the effector remains activated. They are also otherwise called as GTPase –activating proteins (GAPs). The dissociation and association of trimeric subunits of G-Proteins completes one active cycle of G-Protein function and it wan now ready to start new cycle.