CARBOHYDRATES
Carbohydrates are the most abundant class of organic compounds found
in living organisms. They originate as products of photosynthesis, an
endothermic reductive condensation of carbon dioxide requiring light energy and
the pigment chlorophyll.
nCO2
+ nH2O + energy
![]() CnH2nOn + nO2
CnH2nOn + nO2
The formulas of many carbohydrates can be written as carbon hydrates, Cn(H2O)n, hence their name. The carbohydrates are a major source of metabolic energy, both for plants and for animals that depend on plants for food. Aside from the sugars and starches that meet this vital nutritional role, carbohydrates also serve as a structural material (cellulose), a component of the energy transport compound ATP, recognition sites on cell surfaces, and one of three essential components of DNA and RNA. Carbohydrates are called saccharides or, if they are relatively small, sugars. Several classifications of carbohydrates have proven useful, and are outlined in the table.
|
S.No |
Basics |
Types |
|
1. |
Complexity |
Simple Carbohydrates Complex Carbohydrates |
|
2. |
Size |
Tetrose Pentose Hexose Heptose Etc., |
|
3. |
C=O Function |
Aldose Ketose |
|
4. |
Reactivity |
Reducing |
Carbohydrates might be discussed under three different headings namely Monosaccharides, Disaccharides and Polysaccharides.
MONOSACCHARIDES:
Monosaccharides might be explained under aldose and ketose headings. Glucose is one of the fine example for monosaccharides. So, it is described in detail.
Glucose:
Carbohydrates have been given non-systematic names, although the suffix ose is generally used. The most common carbohydrate is glucose (C6H12O6). Applying the terms defined above, glucose is a monosaccharide, an aldohexose and a reducing sugar. The general structure of glucose and many other aldohexoses was established by simple chemical reactions. The following diagram illustrates the kind of evidence considered, although some of the reagents shown here are different from those used by the original scientists.
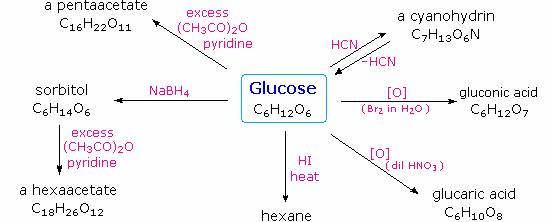
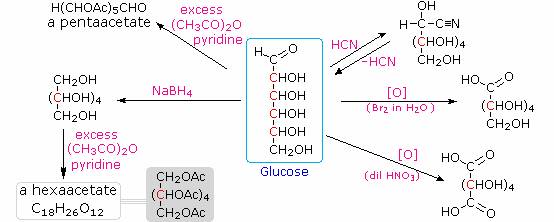
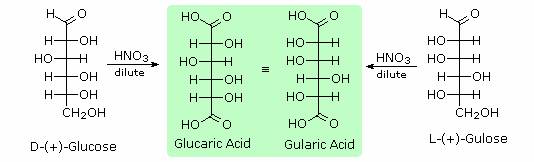
Hot hydriodic acid (HI) was often used to reductively remove oxygen functional groups from a molecule, and in the case of glucose this treatment gave hexane (in low yield). From this it was concluded that the six carbons are in an unbranched chain. The presence of an aldehyde carbonyl group was deduced from cyanohydrin formation, its reduction to the hexa-alcohol sorbitol, also called glucitol, and mild oxidation to the mono-carboxylic acid, glucuronic acid. Somewhat stronger oxidation by dilute nitric acid gave the diacid, glucaric acid, supporting the proposal of a six-carbon chain. The five oxygens remaining in glucose after the aldehyde was accounted for were thought to be in hydroxyl groups, since a penta-acetate derivative could be made. These hydroxyl groups were assigned, one each, to the last five carbon atoms, because geminal hydroxyl groups are normally unstable relative to the carbonyl compound formed by loss of water. Glucose and other saccharides are extensively cleaved by periodic acid, thanks to the abundance of vicinal diol moieties in their structure. This oxidative cleavage, known as the Malaprade reaction is particularly useful for the analysis of selective O-substituted derivatives of saccharides, since ether functions do not react. The stoichiometry of aldohexose cleavage is shown in the following equation.
HOCH2(CHOH)4CHO + 5 HIO4 ——> H2C=O + 5 HCO2H + 5 HIO3
Configuration of Glucose:
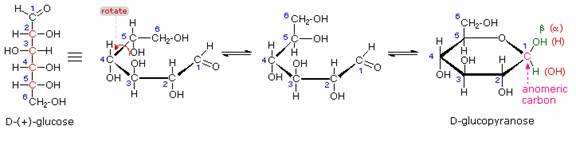
The four chiral centers in glucose indicate there may be as many as sixteen (24) stereoisomers having this constitution. These would exist as eight diastereomeric pairs of enantiomers, and the initial challenge was to determine which of the eight corresponded to glucose. This challenge was accepted and met in 1891 by the German chemist Emil Fischer. His successful negotiation of the stereochemical maze presented by the aldohexoses was a logical tour de force, and it is fitting that he received the 1902 Nobel Prize for chemistry for this accomplishment. One of the first tasks faced by Fischer was to devise a method of representing the configuration of each chiral center in an unambiguous manner. To this end, he invented a simple technique for drawing chains of chiral centers, that we now call the Fischer projection formula.
The problem of drawing three-dimensional configurations on a two-dimensional surface, such as a piece of paper, has been a long-standing concern of chemists. The wedge and hatched line notations we have been using are effective, but can be troublesome when applied to compounds having many chiral centers. As part of his Nobel Prize-winning research on carbohydrates, the great German chemist, Emil Fischer, devised a simple notation that is still widely used. In a Fischer projection drawing, the four bonds to a chiral carbon make a cross with the carbon atom at the intersection of the horizontal and vertical lines. The two horizontal bonds are directed toward the viewer (forward of the stereogenic carbon). The two vertical bonds are directed behind the central carbon (away from the viewer). Since this is not the usual way in which we have viewed such structures, the following diagram shows how a stereogenic carbon positioned in the common two-bonds-in-a-plane orientation ( x–C–y define the reference plane ) is rotated into the Fischer projection orientation (the far right formula). When writing Fischer projection formulas it is important to remember these conventions. Since the vertical bonds extend away from the viewer and the horizontal bonds toward the viewer, a Fischer structure may only be turned by 180º within the plane, thus maintaining this relationship. The structure must not be flipped over or rotated by 90º.
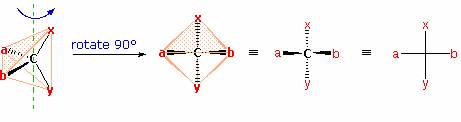
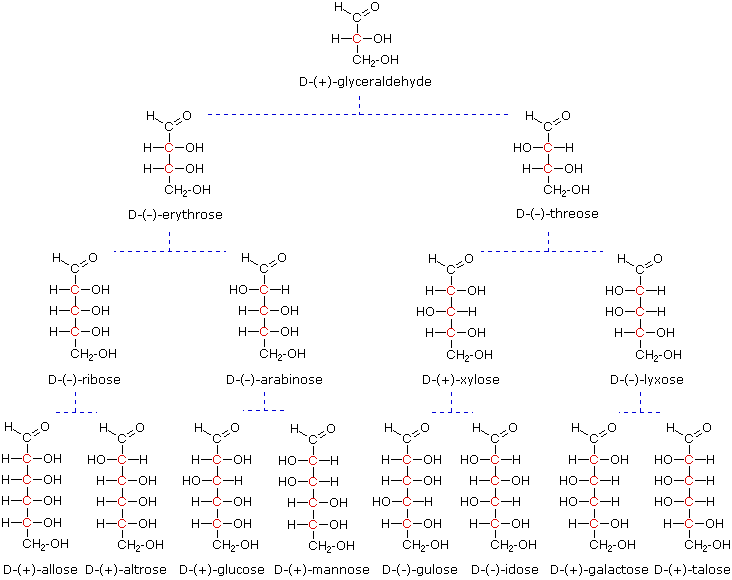
At the time Fischer undertook the glucose project it was not possible to establish the absolute configuration of an enantiomer. Consequently, Fischer made an arbitrary choice for (+)-glucose and established a network of related aldose configurations that he called the D-family. The mirror images of these configurations were then designated the L-family of aldoses. To illustrate using present day knowledge, Fischer projection formulas and names for the D-aldose family (three to six-carbon atoms) are shown below, with the asymmetric carbon atoms (chiral centers) colored red. The last chiral center in an aldose chain (farthest from the aldehyde group) was chosen by Fischer as the D / L designator site. If the hydroxyl group in the projection formula pointed to the right, it was defined as a member of the D-family. A left directed hydroxyl group (the mirror image) then represented the L-family. Fischer's initial assignment of the D-configuration had a 50:50 chance of being right, but all his subsequent conclusions concerning the relative configurations of various aldoses were soundly based. In 1951 x-ray fluorescence studies of (+)-tartaric acid, carried out in the Netherlands by Johannes Martin Bijvoet (pronounced "buy foot"), proved that Fischer's choice was correct. It is important to recognize that the sign of a compound's specific rotation (an experimental number) does not correlate with its configuration (D or L). It is a simple matter to measure an optical rotation with a polarimeter. Determining an absolute configuration usually requires chemical interconversion with known compounds by stereospecific reaction paths.
Anomeric forms of glucose:
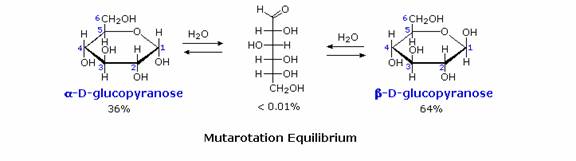
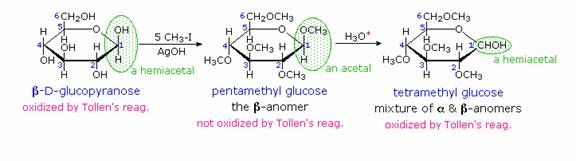
Fischers brilliant elucidation of the configuration of glucose did not remove all uncertainty concerning its structure. Two different crystalline forms of glucose were reported in 1895. Each of these gave all the characteristic reactions of glucose, and when dissolved in water equilibrated to the same mixture. This equilibration takes place over a period of many minutes, and the change in optical activity that occurs is called mutarotation. These facts are summarized in the diagram.

When glucose was converted to its pentamethyl ether (reaction with excess CH3I & AgOH), two different isomers were isolated, and neither exhibited the expected aldehyde reactions. Acid-catalyzed hydrolysis of the pentamethyl ether derivatives, however, gave a tetramethyl derivative that was oxidized by Tollen's reagent and reduced by sodium borohydride, as expected for an aldehyde. These reactions will be displayed.
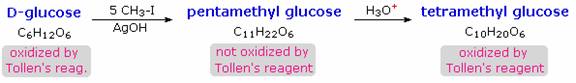
The search for scientific truth often proceeds in stages, and the structural elucidation of glucose serves as a good example. It should be clear from the new evidence presented above, that the open chain pentahydroxyhexanal structure drawn above must be modified. Somehow a new stereogenic center must be created, and the aldehyde must be deactivated in the pentamethyl derivative. A simple solution to this dilemma is achieved by converting the open aldehyde structure for glucose into a cyclic hemiacetal, called a glucopyranose, as shown in the following diagram. The linear aldehyde is tipped on its side, and rotation about the C4-C5 bond brings the C5-hydroxyl function close to the aldehyde carbon. For ease of viewing, the six-membered hemiacetal structure is drawn as a flat hexagon, but it actually assumes a chair conformation. The hemiacetal carbon atom (C-1) becomes a new stereogenic center, commonly referred to as the anomeric carbon, and the α and β-isomers are called anomers.
Consequently, fresh solutions of either alpha or beta-glucose crystals in water should establish an equilibrium mixture of both anomers, plus the open chain chain form. Note that despite the very low concentration of the open chain aldehyde in this mixture, typical chemical reactions of aldehydes take place rapidly. Second, a pentamethyl ether derivative of the pyranose structure converts the hemiacetal function to an acetal.
Acetals are stable to base, so this product should not react with Tollen's reagent or be reduced by sodium borohydride. Acid hydrolysis of acetals regenerates the carbonyl and alcohol components, and in the case of the glucose derivative this will be a tetramethyl ether of the pyranose hemiacetal. This compound will, of course, undergo typical aldehyde reactions.
Cyclic forms of Monosaccharides:
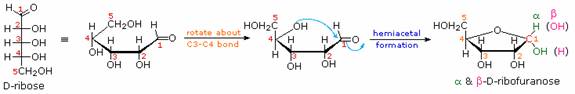
As noted above, the preferred structural form of many monosaccharides may be that of a cyclic hemiacetal. Five and six-membered rings are favored over other ring sizes because of their low angle and eclipsing strain. Cyclic structures of this kind are termed furanose (five-membered) or pyranose (six-membered), reflecting the ring size relationship to the common heterocyclic compounds furan and pyran shown on the right. Ribose, an important aldopentose, commonly adopts a furanose structure, as shown in the following illustration. By convention for the D-family, the five-membered furanose ring is drawn in an edgewise projection with the ring oxygen positioned away from the viewer. The anomeric carbon atom (colored red here) is placed on the right. The upper bond to this carbon is defined as beta, the lower bond then is alpha.
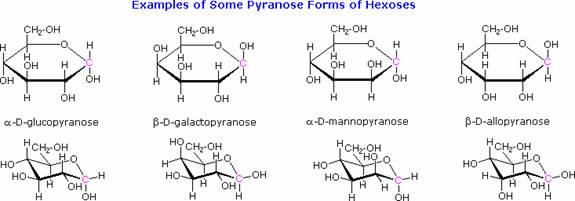
The cyclic pyranose forms of various monosaccharides are often drawn in a flat projection known as a Haworth formula, after the British chemist, Norman Haworth. As with the furanose ring, the anomeric carbon is placed on the right with the ring oxygen to the back of the edgewise view. In the D-family, the alpha and beta bonds have the same orientation defined for the furanose ring. These Haworth formulas are convenient for displaying stereochemical relationships, but do not represent the true shape of the molecules. These molecules are actually puckered in a fashion called as a chair conformation. Examples of four typical pyranose structures are shown below, both as Haworth projections and as the more representative chair conformers.
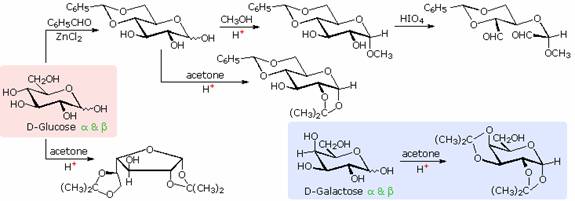
The size of the cyclic hemiacetal ring adopted by a given sugar is not constant, but may vary with substituents and other structural features. Aldolhexoses usually form pyranose rings and their pentose homologs tend to prefer the furanose form, but there are many counter examples. The formation of acetal derivatives illustrates how subtle changes may alter this selectivity. A pyranose structure for D-glucose is drawn in the rose-shaded box on the left. Acetal derivatives have been prepared by acid-catalyzed reactions with benzaldehyde and acetone. As a rule, benzaldehyde forms six-membered cyclic acetals, whereas acetone prefers to form five-membered acetals. The top equation shows the formation and some reactions of the 4,6-O-benzylidene acetal, a commonly employed protective group. A methyl glycoside derivative of this compound (see below) leaves the C-2 and C-3 hydroxyl groups exposed to reactions such as the periodic acid cleavage, shown as the last step. The formation of an isopropylidene acetal at C-1 and C-2, center structure, leaves the C-3 hydroxyl as the only unprotected function. Selective oxidation to a ketone is then possible. Finally, direct di-O-isopropylidiene derivatization of glucose by reaction with excess acetone results in a change to a furanose structure in which the C-3 hydroxyl is again unprotected. However, the same reaction with D-galactose, shown in the blue-shaded box, produces a pyranose product in which the C-6 hydroxyl is unprotected. Both derivatives do not react with Tollens' reagent. This difference in behavior is attributed to the cis-orientation of the C-3 and C-4 hydroxyl groups in galactose, which permits formation of a less strained five-membered cyclic acetal, compared with the trans-C-3 and C-4 hydroxyl groups in glucose. Derivatizations of this kind permit selective reactions to be conducted at different locations in these highly functionalized molecules.
IMPORTANT REACTIONS OF MONOSACCHARIDES:
Oxidation:
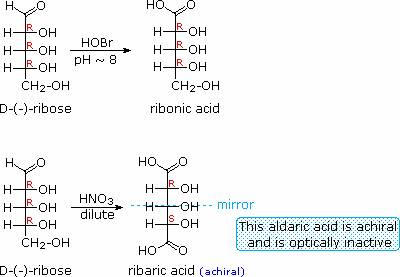
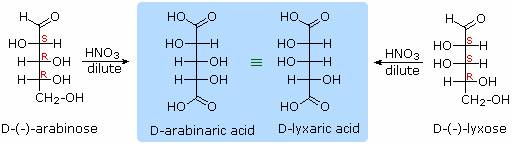
Sugars may be classified as reducing or non-reducing based on their reactivity with Tollens', Benedict's or Fehling's reagents. If a sugar is oxidized by these reagents it is called reducing, since the oxidant (Ag(+) or Cu(+2)) is reduced in the reaction, as evidenced by formation of a silver mirror or precipitation of cuprous oxide. The Tollens' test is commonly used to detect aldehyde functions; and because of the facile interconversion of ketoses and aldoses under the basic conditions of this test, ketoses such as fructose also react and are classified as reducing sugars. When the aldehyde function of an aldose is oxidized to a carboxylic acid the product is called an aldonic acid. Because of the 2º hydroxyl functions that are also present in these compounds, a mild oxidizing agent such as hypobromite must be used for this conversion (equation 1). If both ends of an aldose chain are oxidized to carboxylic acids the product is called an aldaric acid. By converting an aldose to its corresponding aldaric acid derivative, the ends of the chain become identical (this could also be accomplished by reducing the aldehyde to CH2OH, as noted below). Such an operation will disclose any latent symmetry in the remaining molecule. Thus, ribose, xylose, allose and galactose yield achiral aldaric acids which are, of course, not optically active. The ribose oxidation is shown in equation 2. Other aldose sugars may give identical chiral aldaric acid products, implying a unique configurational relationship. The examples of arabinose and lyxose shown in equation 3 above illustrate this result. Remember, a Fischer projection formula may be rotated by 180º in the plane of projection without changing its configuration.
Reduction:
Sodium borohydride reduction of an aldose makes the ends of the resulting alditol chain identical, HOCH2(CHOH)nCH2OH, thereby accomplishing the same configurational change produced by oxidation to an aldaric acid. Thus, allitol and galactitol from reduction of allose and galactose are achiral, and altrose and talose are reduced to the same chiral alditol.
HOBr Oxidation ——> HOCH2(CHOH)nCO2H
an Aldonic Acid
HNO3 Oxidation ——> H2OC(CHOH)nCO2H
an Aldaric Acid
NaBH4
Reduction ——> HOCH2(CHOH)nCH2OH
an Alditol
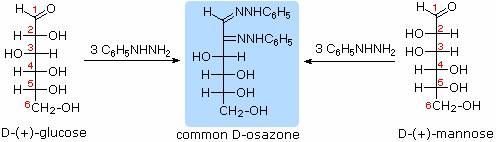
Ozasone formation:
The osazone reaction was developed and used by Emil Fischer to identify aldose sugars differing in configuration only at the alpha-carbon. The upper equation shows the general form of the osazone reaction, which effects an alpha-carbon oxidation with formation of a bis-phenylhydrazone, known as an osazone. Application of the osazone reaction to D-glucose and D-mannose demonstrates that these compounds differ in configuration only at C-2.
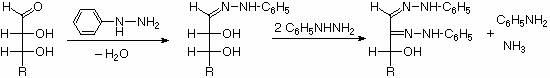
Chain Shortening and Lengthening:
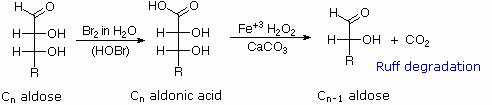
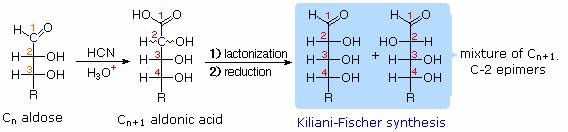
These two procedures permit an aldose of a given size to be related to homologous smaller and larger aldoses. The importance of these relationships may be seen in the array of aldose structures presented earlier, where the structural connections are given by the dashed blue lines. Thus Ruff degradation of the pentose arabinose gives the tetrose erythrose. Working in the opposite direction, a Kiliani-Fischer synthesis applied to arabinose gives a mixture of glucose and mannose.
Using these reactions Fischer's train of logic in assigning the configuration of D-glucose might be obtained.
1. Ribose and arabinose (two well known pentoses) both gave erythrose on Ruff degradation. As expected, Kiliani-Fischer synthesis applied to erythrose gave a mixture of ribose and arabinose.
2. Oxidation of erythrose gave an achiral (optically inactive) aldaric acid. This defines the configuration of erythrose.
3. Oxidation of ribose gave an achiral (optically inactive) aldaric acid. This defines the configuration of both ribose and arabinose.
4. Ruff shortening of glucose gave arabinose, and Kiliani-Fischer synthesis applied to arabinose gave a mixture of glucose and mannose.
5. Glucose and mannose are therefore epimers at C-2, a fact confirmed by the common product from their osazone reactions.
6. A pair of structures for these epimers can be written, but which is glucose and which is mannose?
In order to determine which of these epimers was glucose, Fischer made use of the inherent C2 symmetry in the four-carbon dissymmetric core of one epimer (B). This is shown in the following diagram by a red dot where the symmetry axis passes through the projection formula. Because of this symmetry, if the aldehyde and 1º-alcohol functions at the ends of the chain are exchanged, epimer B would be unchanged; whereas A would be converted to a different compound.
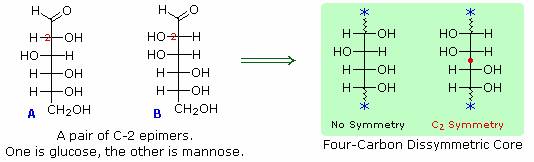
Fischer looked for and discovered a second aldohexose that represented the end group exchange for the epimer lacking the latent C2 symmetry (A). This compound was L-(+)-gulose, and its exchange relationship to D-(+)-glucose was demonstrated be oxidation to a common aldaric acid product. The remaining epimer is therefore mannose.
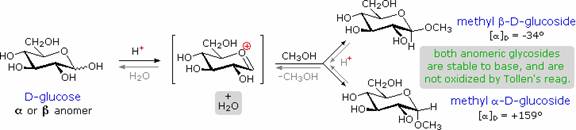
Glycosides:
Acetal derivatives formed when a monosaccharide reacts with an alcohol in the presence of an acid catalyst are called glycosides. This reaction is illustrated for glucose and methanol in the diagram below. In naming of glycosides, the "ose" suffix of the sugar name is replaced by "oside", and the alcohol group name is placed first. As is generally true for most aldols, glycoside formation involves the loss of an equivalent of water. The diether product is stable to base and alkaline oxidants such as Tollen's reagent. Since acid-catalyzed aldolization is reversible, glycosides may be hydrolyzed back to their alcohol and sugar components by aqueous acid. The anomeric methyl glucosides are formed in an equilibrium ratio of 66% alpha to 34% beta. From the structures in the previous diagram, we see that pyranose rings prefer chair conformations in which the largest number of substituents are equatorial. In the case of glucose, the substituents on the beta-anomer are all equatorial, whereas the C-1 substituent in the alpha-anomer changes to axial. Since substituents on cyclohexane rings prefer an equatorial location over axial (methoxycyclohexane is 75% equatorial), the preference for alpha-glycopyranoside formation is unexpected, and is referred to as the anomeric effect.
Glycosides abound in biological systems. By attaching a sugar moiety to a lipid or benzenoid structure, the solubility and other properties of the compound may be changed substantially. Because of the important modifying influence of such derivatization, numerous enzyme systems, known as glycosidases, have evolved for the attachment and removal of sugars from alcohols, phenols and amines. Chemists refer to the sugar component of natural glycosides as the glycon and the alcohol component as the aglycon. Two examples of naturally occuring glycosides and one example of an amino derivative will be displayed in the figure.
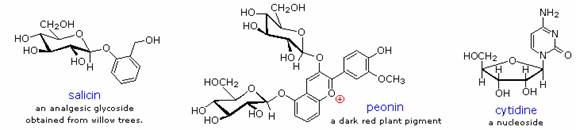
Salicin, one of the oldest herbal remedies known, was the model for the synthetic analgesic aspirin. A large class of hydroxylated, aromatic oxonium cations called anthocyanins provide the red, purple and blue colors of many flowers, fruits and some vegetables. Peonin is one example of this class of natural pigments, which exhibit a pronounced pH color dependence. The oxonium moiety is only stable in acidic environments, and the color changes or disappears when base is added. The complex changes that occur when wine is fermented and stored are in part associated with glycosides of anthocyanins. Finally, amino derivatives of ribose, such as cytidine play important roles in biological phosphorylating agents, coenzymes and information transport and storage materials.
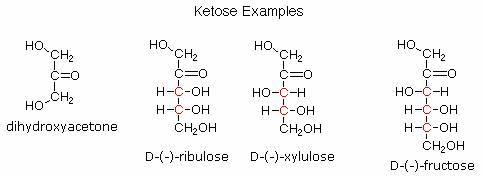
Ketoses:
If a monosaccharide has a carbonyl function on one of the inner atoms of the carbon chain it is classified as a ketose. Dihydroxyacetone may not be a sugar, but it is included as the ketose analog of glyceraldehyde. The carbonyl group is commonly found at C-2, as illustrated by the following examples (chiral centers are colored red). As expected, the carbonyl function of a ketose may be reduced by sodium borohydride, usually to a mixture of epimeric products. D-Fructose, the sweetest of the common natural sugars, is for example reduced to a mixture of D-glucitol (sorbitol) and D-mannitol, named after the aldohexoses from which they may also be obtained by analogous reduction. Mannitol is itself a common natural carbohydrate.
Although the ketoses are distinct isomers of the aldose monosaccharides, the
chemistry of both classes is linked due to their facile interconversion in the
presence of acid or base catalysts. This interconversion, and the corresponding
epimerization at sites alpha to the carbonyl functions, occurs by way of an
enediol tautomeric intermediate. Because of base-catalyzed isomerizations of
this kind, the Tollens' reagent is not useful for distinguishing aldoses from
ketoses or for specific oxidation of aldoses to the corresponding aldonic acids.
Oxidation by HOBr is preferred for the latter conversion.
Functionally and structurally Modified Monosaccharides:
Many modified monosaccharides are deoxy-derivatives. In other words, one or more of the hydroxyl groups present in a normal sugar are missing. Examples of two such deoxy-sugars are given in the diagram.
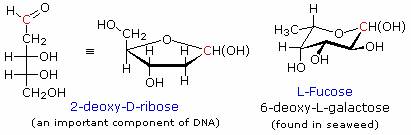
Amino sugars have an amino group, or several amino groups, replacing a customary hydroxyl group. D-Glucosamine, on the left, is probably the most common of the amino sugars. Its N-acetyl amide is the primary monosaccharide unit in the cellulose-like biopolymer that forms the exoskeletons of insects and shellfish.
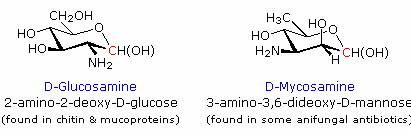
The structure of sialic acid, on the left, can be disconnected to reveal a glucosamine segment bonded to the methyl group of pyruvic acid. Further discussion of the role these saccharide derivatives play in biology is left to more specialized sources.
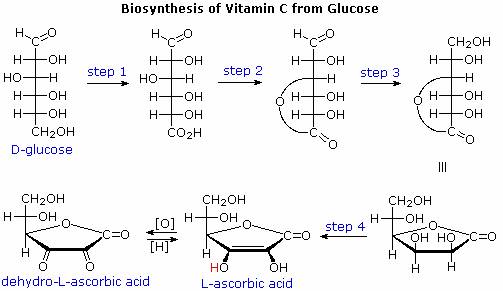
Ascorbic acid, or vitamin C, is an important water soluble biological reducing agent that complements lipid soluble antioxidants such as vitamin E. Most animals synthesize this vital compound from glucose by a series of four enzymatic transformations, shown in the following diagram. Unfortunately, humans and a few other animals, including guinea pigs, some monkeys and many birds, do not have the enzyme, L-gulonolactone oxidase, needed for the fourth step. These animals must consume ascorbic acid as part of their daily diet or suffer chronic deficiency effects, which in extreme cases is the disease called scurvy. Although it does not have a free carboxyl group, ascorbic acid has a pKa of 4.17 as a consequence of vinylagous activation of the C-3 hydroxyl group.
Sweetening Agents:
Sweetness is one of five types
of taste sensed by humans. The others are saliness, sourness, bitterness and
savouriness. It is generally regarded as a pleasureable sensation, and the
simple carbohydrates or sugars that contribute to it are sought after and
valued. Sucrose, from sugar cane and sugar beets, is the standard sweetner used
in western cusine. A less expensive alternative known as high fructose corn
syrup (HFCS) was developed in the late 1950's and is now widely used in baked
goods and beverages. HFCS is made from corn syrup by enzymatic conversion of
glucose to fructose. Since fructose is 2.3 times as sweet as glucose and 75%
sweeter than sucrose, HFCS provides a practical substitute for sucrose in a
variety of applications, and is available in compositions ranging from 45 to 90%
fructose.
Hydrolysis of sucrose produces an equimolar mixture of glucose and fructose,that
is sweeter than sucrose itself. Since the specific rotation of these sugar
solutions changes from +66.5º for pure sucrose to -22.0º for the hydrolysis
mixture (fructose is strongly levorotatory), the resulting glucose fructose
mixture is called invert sugar. It is widely used in food manufacture in much
the same way as HFCS. An enzyme, invertase, which catalyzes the hydrolysis of
sucrose in living organisms, is used in the manufacture of invert sugar. Honey
is similar to invert sugar, consisting roughly of 38% fructose, 31% glucose, 9%
disaccharides such as maltose and 17% water. Although there is a strong
correlation between the rise of obesity in the US and the use of HFCS for
sweetening beverages and foods, it is not clear whether this is a causal
relationship. In fact, no substantial evidence supports the idea that
high-fructose corn syrup is responsible per se for obesity. Instead,
overconsumption of sugars, encouragied by the low cost of HFCS and invert sugar,
is the general culprit.
Insofar as the public is
concerned, the sweet taste of sugars is undoubtedly their most important
characteristic. In this respect, synthetic sweetening agents have become a
multimillion dollar business, thanks to the national preoccupation with weight
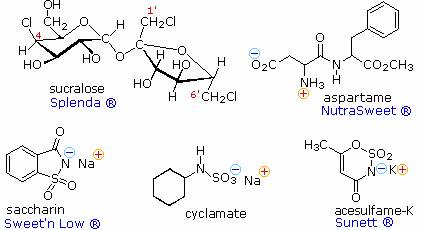
control. The structural formulas for five compounds of this kind are shown on
the right, together with some of the commercial names under which they are sold.
Saccharin was discovered in 1879 at Johns Hopkins University, and is the oldest
member of this group. Sucralose is the newest sweetener, with FDA approval being
issued in 1999.
Because the synthetic sweeteners are many times sweeter than sucrose, only small
amounts are needed to achieve a desired effect. Also, most are not significantly
metabolized, so their use does not introduce additional calories into a diet.
For some individuals, however, taste overtones such as bitterness reduce the
suitability of these agents as sugar substitutes. If the sweetness of sucrose is
taken as a standard, then these and other sweetening agents may be ranked
accordingly as shown in the table.
|
Compounds |
sucralose | saccharin | acesulfame-K | aspartame | cyclamate | fructose | sucrose | glucose | maltose | lactose |
|
Sweetness |
600 |
300 |
200 |
180 |
30 |
1.7 |
1.0 |
0.7 |
0.3 |
0.15 |
Aspartame is a dipeptide composed of two natural amino acids, phenylalanine and aspartic acid, neither of which is sweet. Each of these components has a stereogenic center, so four stereoisomers are possible. The natural configuration of these amino acids is 2S, and (S,S)-Aspartame is the commercial sweetener. The other three stereoisomers are not sweet, and one is bitter. Aspartame undergoes a slow intramolecular acylation to a cyclic dilactam that is not sweet. Consequently, soft drinks and other beverages that contain dissolved aspertame have a limited shelf life. This slow reaction is accelerated by heat, so aspertame is not suitable for cooking purposes.
Acesulfame, cyclamate and saccharin are achiral, and are generally suitable for cooking Since these compounds are acidic, their water soluble sodium or potassium salts are the commonly used form. Sucralose has many stereogenic centers, and although other chloro derivatives of sucrose are also sweet, only the designated stereoisomer has been approved as a food additive.

Stevia is a genus of herbs and
shrubs native to subtropical and tropical South America and Central America. The
leaves of the plant Stevia rebaudiana Bertoni have a sweet taste
resulting from glycosides of the diterpene steviol (structure on the right).
Stevioside and rebaudioside A, the primary components, are glucosides attached
to the hydroxyl functions of steviol. They are heat stable, pH stable, and do
not ferment. Stevioside has a sweetness 200 to 350 times that of sucrose, and a
relative caloric value 300 fold less. Since stevioside does not induce a
glycemic response when ingested, it offers potential as a natural sweetener for
diabetics and others on carbohydrate-controlled diets.
In South America, stevia leaves have been employed in ethnomedical applications
for centuries. In Japan, stevia extracts have been used as a sweetner for over
thirty years with no reported harmful effects. Nevertheless, in 1991, at the
request of an anonymous complaint, the United States Food and Drug
Administration (FDA) labeled stevia as an "unsafe food additive" and restricted
its import. The FDA's stated reason was "toxicological information on stevia is
inadequate to demonstrate its safety." The 1994 Dietary Supplement Health and
Education Act forced the FDA in 1995 to revise its stance to permit stevia to be
used as a dietary supplement, although not as a food additive – a position that
seems contradictory because it simultaneously labels stevia as safe and unsafe,
depending on how it is sold. Additional studies have shown that stevia improves
insulin sensitivity in rats, possibly promoting insulin production. Also,
preliminary human studies suggest that stevia may help reduce hypertension.
Despite other research pointing to the safety of stevia, government agencies
continue to express concern over a lack of conclusive evidence on this subject.
DISACCHARIDES:
When the alcohol component of a glycoside is provided by a hydroxyl function on another monosaccharide, the compound is called a disaccharide. Four examples of disaccharides composed of two glucose units are shown in the following diagram. The individual glucopyranose rings are labled A and B, and the glycoside bonding is circled in light blue. Notice that the glycoside bond may be alpha, as in maltose and trehalose, or beta as in cellobiose and gentiobiose. Acid-catalyzed hydrolysis of these disaccharides yields glucose as the only product. Enzyme-catalyzed hydrolysis is selective for a specific glycoside bond, so an alpha-glycosidase cleaves maltose and trehalose to glucose, but does not cleave cellobiose or gentiobiose. A beta-glycosidase has the opposite activity. In order to draw a representative structure for cellobiose, one of the glucopyranose rings must be rotated by 180º, but this feature is often omitted in favor of retaining the usual perspective for the individual rings. The bonding between the glucopyranose rings in cellobiose and maltose is from the anomeric carbon in ring A to the C-4 hydroxyl group on ring B. This leaves the anomeric carbon in ring B free, so cellobiose and maltose both may assume alpha and beta anomers at that site (the beta form is shown in the diagram). Gentiobiose has a beta-glycoside link, originating at C-1 in ring A and terminating at C-6 in ring B. Its alpha-anomer is drawn in the diagram. Because cellobiose, maltose and gentiobiose are hemiacetals they are all reducing sugars (oxidized by Tollen's reagent). Trehalose, a disaccharide found in certain mushrooms, is a bis-acetal, and is therefore a non-reducing sugar. A systematic nomenclature for disaccharides exists, but as the following examples illustrate, these are often lengthy.
Cellobiose : 4-O-β-D-Glucopyranosyl-D-glucose (the
beta-anomer is drawn)
Maltose : 4-O-α-D-Glucopyranosyl-D-glucose (the beta-anomer is drawn)
Gentiobiose : 6-O-β-D-Glucopyranosyl-D-glucose (the alpha-anomer is
drawn)
Trehalose : α-D-Glucopyranosyl-α-D-glucopyranoside
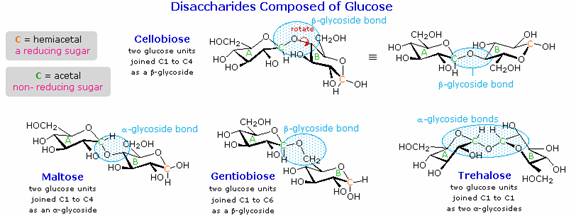
Although all the disaccharides shown here are made up of two glucopyranose rings, their properties differ in interesting ways. Maltose, sometimes called malt sugar, comes from the hydrolysis of starch. It is about one third as sweet as cane sugar (sucrose), is easily digested by humans, and is fermented by yeast. Cellobiose is obtained by the hydrolysis of cellulose. It has virtually no taste, is indigestible by humans, and is not fermented by yeast. Some bacteria have beta-glucosidase enzymes that hydrolyze the glycosidic bonds in cellobiose and cellulose. The presence of such bacteria in the digestive tracts of cows and termites permits these animals to use cellulose as a food. Finally, it may be noted that trehalose has a distinctly sweet taste, but gentiobiose is bitter.
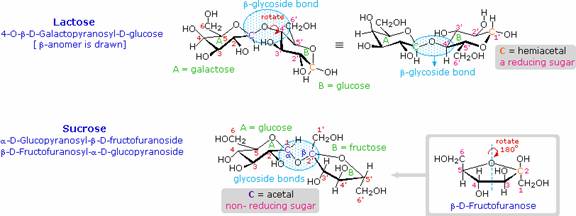
Disaccharides made up of other sugars are known, but glucose is often one of the components. Lactose and Sucrose are examples of such mixed disaccharides. Lactose, also known as milk sugar, is a galactose-glucose compound joined as a beta-glycoside. It is a reducing sugar because of the hemiacetal function remaining in the glucose moiety. Many adults, particulaly those from regions where milk is not a dietary staple, have a metabolic intolerance for lactose. Infants have a digestive enzyme which cleaves the beta-glycoside bond in lactose, but production of this enzyme stops with weaning. Cheese is less subject to the lactose intolerance problem, since most of the lactose is removed with the whey. Sucrose, or cane sugar, is our most commonly used sweetening agent. It is a non-reducing disaccharide composed of glucose and fructose joined at the anomeric carbon of each by glycoside bonds (one alpha and one beta). In the formula shown here the fructose ring has been rotated 180º from its conventional perspective.
TRI AND HIGH OLIGOSACCHARIDES:
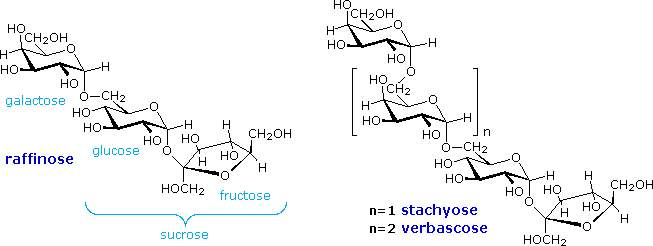
Complex oligosaccharides are common components of numerous biologically important macromolecules. In many of these systems aminosaccharides, deoxysaccharides and C9 glyconic acids are found linked to more common sugar units, so an amazing diversity of similar but distinct structures exists. In this discussion we shall limit our attention to relatively simple molecules composed of simple aldohexose units. Appreciable amounts of oligosaccharides are found in certain foods, such as peas and beans. The structural formulas of three such compounds are given in the following diagram. They are all non-reducing sugars. A common sucrose moiety is seen for the two rings on the right, and this is joined to one or more galactopyranose rings by alpha-glycoside bonds at C-6. Two enzymes are rquired to hydrolyze these oligosaccharides into monosaccharides that are easily absorbed into the blood stream. The galactose units are cleaved by alpha-galactosidase, and the glucose-fructose link in sucrose is hydrolyzed by sucrase (or invertase). Humans do not have a source of alpha-galactosidase in their digestive system, so the oligosaccharide passes largely unchanged into the colon. Anerobic microorganisms in the colon ferment these sugars, producing carbon dioxide and methane, gases that cause flatulence.
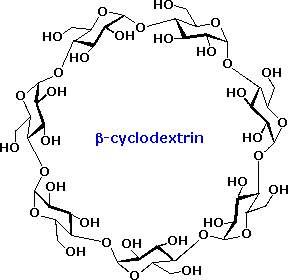
An interesting class of non-reducing oligosaccharides composed of glucopyranose rings joined 1-4 by alpha-glycosidic bonds are called cyclodextrins. Cyclodextrins are formed when starch is treated with an amylase enzyme from Bacillus macerans. Depending on the number of glucose units in the ring the cyclodextrins are named alpha (6), beta (7), and gamma (8). The shape of the cyclodextrans is that of a tapered ring, with the C-2 and C-3 hydroxyl functions on one edge and the CH2OH groups hanging from the opposite edge. The structure of the beta-isomer is shown on the right. Because the interior of the cyclodextrin ring is relatively hydrophobic, these remarkable compounds are able to encapsulate small nonpolar molecules. They have been used as catalysts and aqueous transport agents.
POLYSACCHARIDES:
Polysaccharides are large high-molecular weight molecules constructed by joining monosaccharide units together by glycosidic bonds. They are sometimes called glycans. The most important compounds in this class, cellulose, starch and glycogen are all polymers of glucose. This is easily demonstrated by acid-catalyzed hydrolysis to the monosaccharide. Since partial hydrolysis of cellulose gives varying amounts of cellobiose, we conclude the glucose units in this macromolecule are joined by beta-glycoside bonds between C-1 and C-4 sites of adjacent sugars. Partial hydrolysis of starch and glycogen produces the disaccharide maltose together with low molecular weight dextrans, polysaccharides in which glucose molecules are joined by alpha-glycoside links between C-1 and C-6, as well as the alpha C-1 to C-4 links found in maltose. Polysaccharides built from other monosaccharides (e.g. mannose, galactose, xylose and arabinose) are also known.
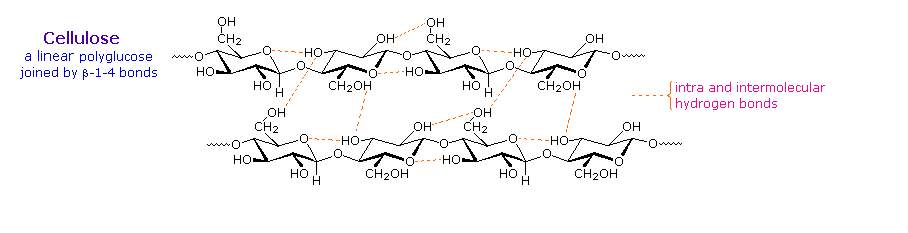
Over half of the total organic carbon in the earth's biosphere is in cellulose. Cotton fibres are essentially pure cellulose, and the wood of bushes and trees is about 50% cellulose. As a polymer of glucose, cellulose has the formula (C6H10O5)n where n ranges from 500 to 5,000, depending on the source of the polymer. The glucose units in cellulose are linked in a linear fashion, as shown in the drawing below. The beta-glycoside bonds permit these chains to stretch out, and this conformation is stabilized by intramolecular hydrogen bonds. A parallel orientation of adjacent chains is also favored by intermolecular hydrogen bonds. Although an individual hydrogen bond is relatively weak, many such bonds acting together can impart great stability to certain conformations of large molecules. Most animals cannot digest cellulose as a food, and in the diets of humans this part of our vegetable intake functions as roughage and is eliminated largely unchanged. Some animals (the cow and termites, for example) harbour intestinal microorganisms that breakdown cellulose into monosaccharide nutrients by the use of beta-glycosidase enzymes. Cellulose is commonly accompanied by a lower molecular weight, branched, amorphous polymer called hemicellulose. In contrast to cellulose, hemicellulose is structurally weak and is easily hydrolyzed by dilute acid or base. Also, many enzymes catalyze its hydrolysis. Hemicelluloses are composed of many D-pentose sugars, with xylose being the major component. Mannose and mannuronic acid are often present, as well as galactose and galacturonic acid.
Cotton, probably the most useful natural fiber, is nearly pure cellulose. The manufacturie of textiles from cotton involves physical manipulation of the raw material by carding, combing and spinning selected fibers. For fabrics the best cotton has long fibers, and short fibers or cotton dust are removed. Crude cellulose is also available from wood pulp by dissolving the lignan matrix surrounding it. These less desirable cellulose sources are widely used for making paper. In order to expand the ways in which cellulose can be put to practical use, chemists have devised techniques for preparing solutions of cellulose derivatives that can be spun into fibers, spread into a film or cast in various solid forms. A key factor in these transformations are the three free hydroxyl groups on each glucose unit in the cellulose chain, --[C6H7O(OH)3]n--. Esterification of these functions leads to polymeric products having very different properties compared with cellulose itself.
Cellulose Nitrate, first prepared over 150 years ago by treating cellulose with nitric acid, is the earliest synthetic polymer to see general use. The fully nitrated compound, --[C6H7O(ONO2)3]n--, called guncotton, is explosively flammable and is a component of smokeless powder. Partially nitrated cellulose is called pyroxylin. Pyroxylin is soluble in ether and at one time was used for photographic film and lacquers. The high flammability of pyroxylin caused many tragic cinema fires during its period of use. Furthermore, slow hydrolysis of pyroxylin yields nitric acid, a process that contributes to the deterioration of early motion picture films in storage.
Cellulose Acetate, --[C6H7O(OAc)3]n--, is less flammable than pyroxylin, and has
replaced it in most applications. It is prepared by reaction of cellulose with
acetic anhydride and an acid catalyst. The properties of the product vary with
the degree of acetylation. Some chain shortening occurs unavoidably in the
preparations. An acetone solution of cellulose acetate may be forced through a
spinnerette to generate filaments, called acetate rayon, that can be woven into
fabrics.
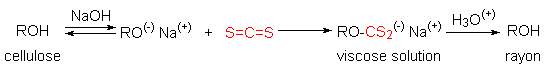
Viscose Rayon, is prepared by formation of an alkali soluble xanthate derivative
that can be spun into a fiber that reforms the cellulose polymer by acid
quenching. The following general equation illustrates these transformations. The
product fiber is called viscose rayon.
Starch is a polymer of glucose,
found in roots, rhizomes, seeds, stems, tubers and corms of plants, as
microscopic granules having characteristic shapes and sizes. Most animals,
including humans, depend on these plant starches for nourishment. The structure
of starch is more complex than that of cellulose. The intact granules are
insoluble in cold water, but grinding or swelling them in warm water causes them
to burst.
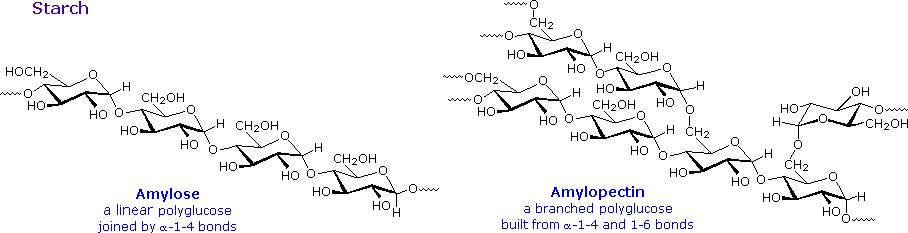
The released starch consists of two fractions. About 20% is a water soluble
material called amylose. Molecules of amylose are linear chains of several
thousand glucose units joined by alpha C-1 to C-4 glycoside bonds. Amylose
solutions are actually dispersions of hydrated helical micelles. The majority of
the starch is a much higher molecular weight substance, consisting of nearly a
million glucose units, and called amylopectin. Molecules of amylopectin are
branched networks built from C-1 to C-4 and C-1 to C-6 glycoside links, and are
essentially water insoluble.
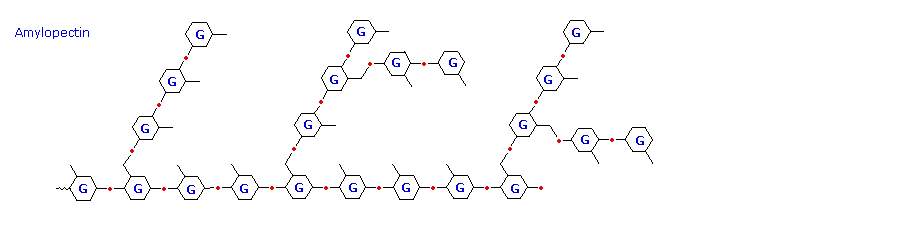
The branching in this diagram is exaggerated, since on average, branches only occur every twenty five glucose units. Hydrolysis of starch, usually by enzymatic reactions, produces a syrupy liquid consisting largely of glucose. When cornstarch is the feedstock, this product is known as corn syrup. It is widely used to soften texture, add volume, prohibit crystallization and enhance the flavor of foods. Glycogen is the glucose storage polymer used by animals. It has a structure similar to amylopectin, but is even more highly branched (about every tenth glucose unit). The degree of branching in these polysaccharides may be measured by enzymatic or chemical analysis.
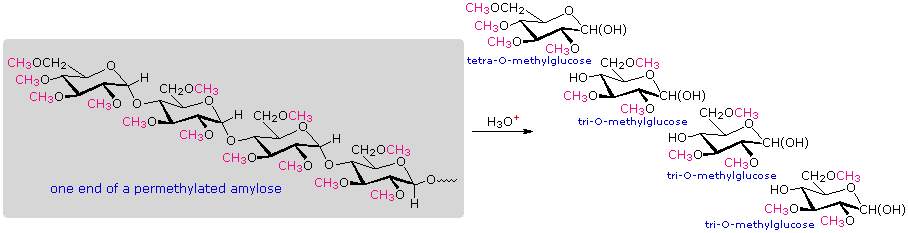
Measuring Chain Branching in Polysaccharides:
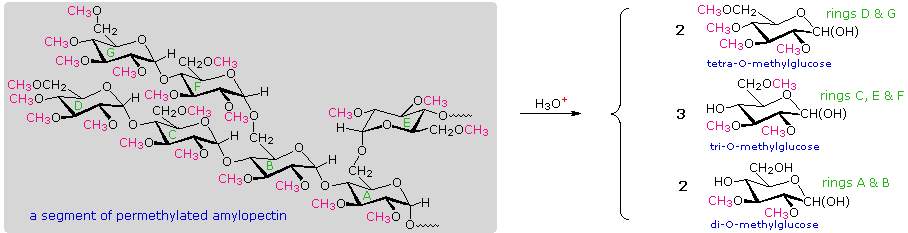
A simple strategy that provides easily interpreted results consists of methylating all the free hydroxyl groups in a polysaccharide, followed by hydrolysis of the glycoside bonds and determination of the methylated glucose products finally obtained. In the following diagram, the methyl groups added to an amylose chain are colored magenta. A chain of glucose units joined only by C-1 to C-4 glycoside bonds will give 2,3,6-tri-O-methylglucose as the chief product with a small amount of 2,3,4,6-tetra-O-methylglucose coming from the glucose unit on the chain's end. The shorter the average chain length, the greater is the relative amount of the tetramethyl derivative.
When chain branching of the kind found in amylopectin and glycogen is prevalent, 2,3-di-O-methylglucose is formed from each glucose at a branch point. The amount of this derivative formed is roughly the same as the tetramethyl derivative, and reflects the degree of chain branching. A segment of permethylated amylopectin will be analyzed. The numbers presented in the diagram reflect that example. Typical results for amylopectin are 90% trimethylglucose + 5% dimethylglucose + 5% tetramethylglucose.