DNA DAMAGE AND REPAIR
DNA molecules, like all other biomolecules, can be damaged in numerous ways. Spontaneous damage due to replication errors, deamination, depurination and oxidation is compounded in the real world by the additional effects of radiation and environmental chemicals. The number of ways that DNA molecules can be damaged is very large. Since repair systems must be capable of recognizing and dealing with each type of damage, it is not surprising that there are a large number of different types of repair system.
Mutation, Mutagen, Mutagenesis and Mutant:
Any change in DNA base sequence referred as Mutation. Agent, which causes mutation, referred as Mutagen. The process through which, mutation occurs known as Mutagenesis. Mutated genetic state of organisms is referred as Mutant. For example In E.coli Lac+ is wild type whereas Lac- is called as mutant.
Mutagens:
A Spontaneous mutation occurs once in 108 cells. The mutation rate can be increase by exposing cells to mutagens, which are either chemicals or physical agents such as UV-irradiation. Both chemicals and physical agents act by causing genetic damage that result in base changes in DNA. Depending upon the nature of mutagens they are classified into six types:
1. Base analogue
2. Chemical agents
3. Intercalating agents
4. Mutator genes
5. Physical agents
6. Biological agents
1. Base analogue
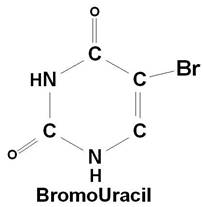
If a mutagen similar to one of the four bases required for the synthesis of DNA then the agents are known as base analogue. They substitutes for a standard base during replication and causes a new base pair to appear in daughter cells in a later generation. For example: 5- Bromo Uracil substituted instead of T or C which intern result in the conversion of A: T --> G: C and G: C --> A: T.
The base analogues used in Mutagenesis are substances that are sufficiently similar to naturally occurring DNA bases so that their deoxyribonucleotide triphosphates can be incorporated into DNA in place of the normal bases. However, they also have anomolous base-pairing properties, leading to an increased rate of mutagenesis. For example, 5-bromouracil pairs like thymine (5-methyluracil), but undergo more enol tautomerization, leading to more frequent mispairing with guanine. Similarly, 2-aminopurine normally pairs with thymine, but can also pair with cytosine. These mispairings lead to an increase in the frequency of transitions.
2. Chemical mutagens:
A chemical mutagen is a substance that can alter a base that is already incorporated in DNA and thereby change its hydrogen-bonding specificity. Three commonly used chemical mutagens include nitrous acid, hydroxylamine and ethyl methane sulfonate.
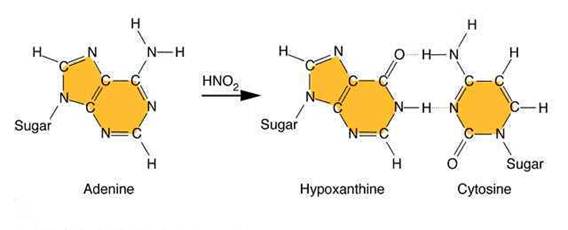
Nitrous acid: Treatment of DNA with nitrous acid leads to deamination of cytosine and adenine, again resulting in transitions.
Hydroxylating agents: Hydroxylamine adds a hydroxyl group to the amino group at position 4 of cytosine, causing it to pair with A instead of G. Hydroxylamine thus causes a very specific CG to TA transition.
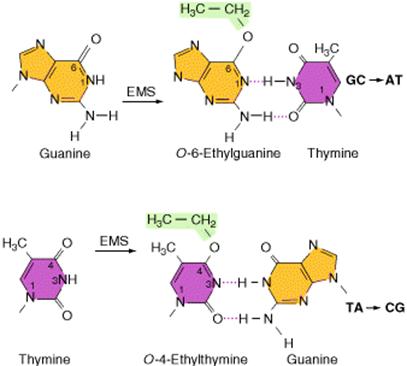
Alkylating agents: Certain alkylating agents, such as ethyl methane sulfonate (
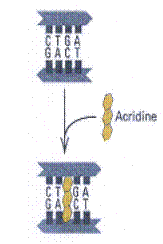
3. Intercalating agents:
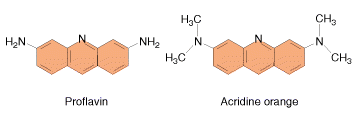
These agents are planar (flat) in structure and are approximately the same size as a purine-pyrimidine base pair. In solution they can insert between stacked base pairs. The best known such agent is Ethidium bromide which is used to visualize DNA, as when it is inserted between the stacked base-pairs it fluoresces brightly to allow the DNA to be visualized under UV radiation. Replication of DNA containing intercalating agents is often seen to result in the addition of single bases, which seriously affects the reading frame of the gene. Proflavin and acridine orange are other intercalating agents. Their activity is because of planar ring structure.
4. Mutator genes:
These are the certain genes, which are usually plays vital role in the prevention of mutation. Defect in these genes might lead to the increased rate of mutation. Such genes are usually referred as mutator genes. Example for such genes include DNA methylase gene, which actively involved in mismatch repair system.
5. Physical agents:
Among the physical agents, UV radiation and Ionizing radiation play vital role in mutation.
i) UV radiations:
Ultraviolet light generates a number of photoproducts in DNA. Two different lesions that occur at adjacent pyrimidine residues include the cyclobutane pyrimidine photodimer (thymidine dimer) and the 6-4 photoproduct.
These lesions interfere with normal base pairing. These changes induce transitional, transversional and frameshift mutations indirectly.
Ultraviolet light is absorbed by the nucleic acid bases, and the resulting influx of energy can induce chemical changes. The most frequent photoproducts are the consequences of bond formation between adjacent pyrimidines within one strand, and, of these, the most frequent are cyclobutane pyrimidine dimers (CPDs). T-T CPDs are formed most readily, followed by T-C or C-T; C-C dimers are least abundant. One can obtain an idea of the extent of distortion of DNA chain structure caused by CPDs by noting that, in the diagram of a T-T CPD below, the cyclobutane ring, should have sides of approximately equal length. Thus the two adjacent pyrimidines must be pulled closer to each other than in normal DNA.
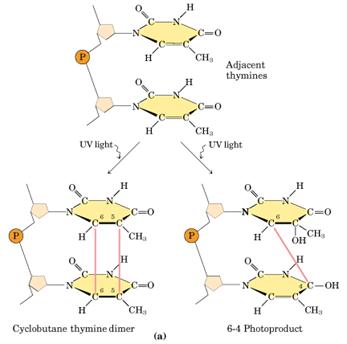
Dimers can also be produced by formation of a single covalent bond between the 6 position of one pyrimidine and the 4 position of the adjacent pyrimidine on the 3' side. The order of abundance of such pyrimidine (6-4) pyrimidine photoproducts (6-4PPs) is T-C>>C-C>T-T>C-T. Although only one bond attaches the adjacent pyrimidines, there is nevertheless extensive distortion of the normal DNA structure.
ii) Ionizing radiations:
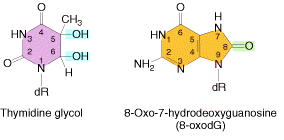
Ionizing radiation like x-rays and gamma rays, results in the formation of ionized and excited molecules that can cause damage to cellular components and to DNA. Because of the aqueous nature of biological systems, the molecules generated by the effects of ionizing radiation on water produce the most damage. Different types of oxygen radicals are produced. These include `OH, O2- and H2O2. These species can damage bases and cause different adducts and degradation products. Among these products, there are two products occurs mostly namely thymine glycol and 8-Oxodeoxy Guanosine.
6. Biological Agents:
Usually viruses placed under this category. Both DNA and RNA viruses found to have the
ability to cause cancer by means of causing mutation i.e. change the DNA
composition of the cell. The main mechanism
through which these viruses achieve their function is that by converting proto-oncogenes
of host cells into oncogenes. By their
action to convert protooncogene to oncogene considered as mutating role of the
viruses. Converted Oncogene found to be
the main factor responsible for the occurrence of cancer in host. For example: Papilloma viruses, Epstein-Barr
virus and Simian Virus (SV) 40 are DNA Viruses.
Rous sarcoma virus and Mouse Mammary Tumor Virus (MMTV) are RNA
Viruses.
TYPES
OF MUTATION:
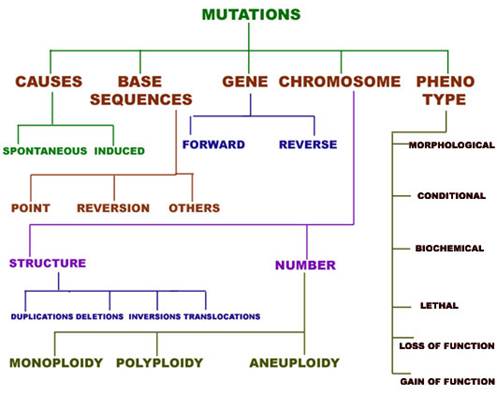
Mutation classified into different types depending upon several factors.
1. Depending upon the nature of occurrence, mutation are of two types namely Spontaneous mutation and Induced mutation. Spontaneous mutation occurs naturally without the involvement of any mutagen whereas if mutation occurs with the involvement of mutagen then they referred as Induced mutation.
1.1. SPONTANEOUS MUTATIONS:
Spontaneous mutations occur mainly due to three reasons namely errors in replication, spontaneous lesions and transposable elements.
1.1. a. Errors in replication:
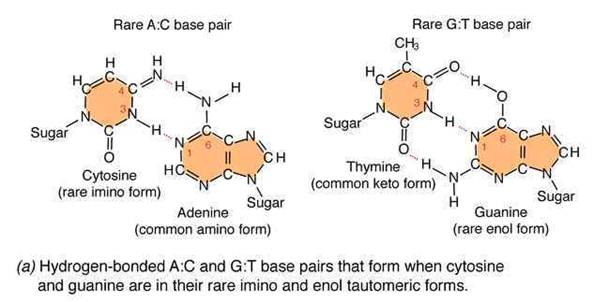
An error in DNA replication can occur when an illegitimate nucleotide pair forms during DNA replication. It intern leads to a base substitution. The main reason for the error in replication is due to the tautomeric shift of bases. Usually bases in DNA occur in many forms, which differ in the positions of their atoms and in the bonds between the atoms. These forms are in equilibrium. The keto form of each base is normally present in DNA, whereas the imino and enol forms of the bases are rare. Different forms of the bases listed below:
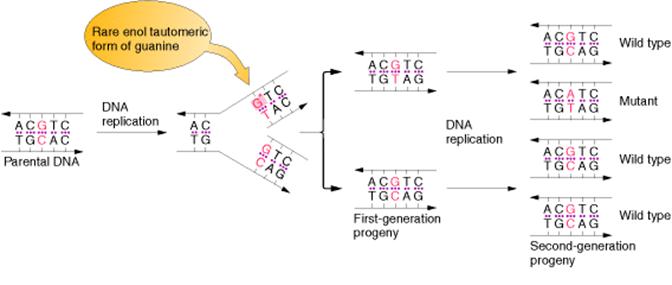
Due to the variation of bases from the usual form to rare form leads to mispairing in DNA. For example when cytosine changed into rare imino form then it is paired with Adenine. Similarly rare enol form of guanine makes it to pair with thymine. These types of changes referred as tautomeric shift. These types of changes intern leads to transitional, transversional and frameshift mutations in DNA.
1.1.b. Spontaneous Lesions:
It is also another type of spontaneous mutation. It occurs mainly in two ways namely depurination and deamination.
1.1. b.1. Depurination:
It is the most common method of spontaneous Lesions. It occurs due to the breakage of glycosidic bond between the base and deoxyribose residues. Due to this, purine bases lost from the DNA. The frequency of depurination is one in 10,000 purines in DNA. Prolonged lesions might leads to severe genetic damages because the resulting apurinic sites cannot specify a base complementary to the original purine.
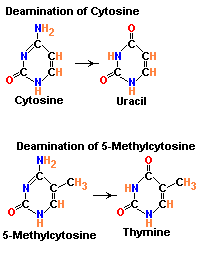
1.1. b.2. Deamination:
Deamination refers to the removal of amino group from bases. Usually cytosine and 5-methyl cytosine undergoes deamination, which result in the production of uracil and thymine respectively. Conversion of cytosine to uracil results in the transitional mutation GC --> AT similarly conversion of 5-methyl cytosine to thymine results in the transitional mutation GC --> AT.
1.1. c. Transposable Elements:
They are also otherwise known as "jumping genes" or Transposans. Due to the transfer of these components into the interior region of structural genes and regulatory genes leads to the abnormal expression of these genes. Thus these elements cause mutations.
1.2 INDUCED MUTATIONS:
This type of mutation is the major type. Mutation induced by the mutagen is found to be example for this type of mutation.
2. Depending upon the change in base sequence, mutation classified into different types namely, Point mutation, translocation, inversion and reversion.
2.1. Point mutation:
When only one base changes then the mutation referred as point mutation. This intern is of three types namely base substitution, base deletion and base insertion.
2.1.1. Base substitution:
Base substitution refers to the change of an single base in gene. It is of two types namely transition and transversion. When purine is replaced by another purine or pyrimidine is replaced by another pyrimidine then it is named as transition. When purine replaced by a pyrimidine and vice versa then it is referred as transversion.
2.1.2. Base deletion:
Removal of Single base from the gene referred as base deletion.
2.1.3. Base addition:
Addition of the single base to the gene referred as base addition.
2.2. Other mutations:
Mutation can also occur with more than one base change like Deletion, Duplication, Inversion, Insertion and Translocation where fragment of DNA undergo changes.
Deletion refers to lose of fragment of DNA. Duplication refers to the doubling of fragment of DNA or repetition of specific DNA fragment two times. Inversion refers to change the polarity of fragment of DNA i.e. fragment of DNA removed and inverted then ligated to its original location. Insertion refers to the addition of fragment of DNA. Translocation refers to the transfer of fragment of DNA from its original location to new location.
2.3. Reversion:
Reversion refers to the type of mutation where the wild type of the genotype is regained due to mutation. It is otherwise known as back mutation or reverse mutation. If second mutation in a gene suppresses the effect of first mutation then this type of mutations called as second site mutations or suppressor mutations. Reversion further divided into two types namely intergenic and intragenic reversion. The difference is mainly because of the position where second site mutation occurs. If it occurs in the same gene then it is called as intragenic reversion and if it occurs in other gene then it is called as intergenic reversion.
Depending upon the survival nature of the organism, mutation found to be of two types namely lethal and leaky mutations. If mutation prevents the growth of the organism then the mutation said to be of lethal mutation type and if mutation reduces the growth rate of the organisms then the muation said to be of leaky mutation type.
3. GENE MUTATION:
Gene mutations results from the changes within structure of gene. It is sometimes called as a point mutation by considering that the change occurs at particular gene or point or loci in chromosome. Because of this mutation type, alleles change. Gene mutation broadly classified into two types depending upon the change with respect to wild type, namely forward mutation and reverse mutation.
When change can converts wild type gene into mutant type then mutation referred as forward mutation whereas change converts mutant type into wild type then mutation referred as reverse or reversion or back mutation.
3.1. FORWARD MUTATION:
Forward mutation studied under two different headings namely
3.1.1. Single nucleotide pair (base pair) substitutions
3.1.2. Single nucleotide pair (base pair) additions and deletions
3.1.1. Single nucleotide pair (base pair) substitutions:
This mutation type studied under two different levels depending upon the molecule's nature namely at DNA level and Protein level. At DNA level, if purine or pyrimidine replaced by a different purine or pyrimidine respectively then the change referred as transition.
AT ------> GC GC ------> AT CG---------->TA TA------>CG
If purine replaced by another pyrimidine and vice versa then the change referred as transversion.
AT---------->CG AT---------->TA GC---------->TA GC ----------->CG
TA---------->GC TA---------->AT CG----------->AT CG---------->GC
At Protein level, depending upon the change occurring in the aminoacid nature in protein mutation studied under three different headings namely silent, missense and nonsense mutation.
If the mutation changes one codon for an aminoacid into another codon for that same amino acid then the mutation referred as silent mutation. In such mutation, there is no change occurs in structure and function of protein.
AGG ----------> CGG
This change of codon does not change amino acid because both codes for the same amino acid, arginine.
Due to mutation, if one amino acid is substituted by another amino acid then it is called as missense mutation. It occurs when codon for one amino acid is replaced by a codon for another amino acid. If a missense mutation causes the substitution of a chemically similar amino acid then the mutation is referred as synonymous substitution or mutation. This type of mutation results in a less severe effect on the protein's structure and function.
AAA---------->AGA
This results in a change of basic amino acid Lysine into another basic amino acid Arginine.
If a missense mutation causes the substitution of a chemically different amino acid then the mutation is called as non-synonymous substitution or mutation. It produces severe changes in protein structure and function. If such mutation occurs in amino acids contributing active site of protein then they are known as null (nothing) mutations.
AAA----------->GAA
This results in a change of basic amino acid Lysine into acidic amino acid Glutamic acid.
If codon for one amino acid is replaced by a translation termination codon or stop or nonsense codon then the mutation was known as nonsense mutation. Non-sense mutations will lead to the premature termination of translation. Thus, they have a considerable effect on protein function. Typically, unless it is very close to the 3' end of the open reading frame so that only a partly functional truncated polypeptide is produced; a nonsense mutation will produce a completely inactive protein product.
CAG ----------> UAG
This results in a change of amino acid, glutamine to a termination codon.
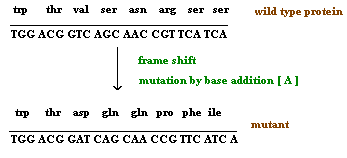
3.1.2. Single nucleotide pair (base pair) additions and deletions:
When a single nucleotide base is added or deleted, it would result in a change in amino acid sequence in protein after the site of mutation. This is because the sequence of mRNA is read by the translational apparatus in groups of three base pairs (codons). Hence these lesions are called frame shift mutations.
These mutations cause the entire aminoacid sequence translationally downstream of the mutant site to bear no relation to the original amino acid sequence. Thus, frameshift mutations typically exhibit complete loss of normal protein structure and function.
3.2. REVERSE MUTATION:
Reverse mutation classified into two types’ namely First site reversion and Second site reversion.
3.2.1. First site Reversion:
If reversion occurs at the same site i.e. in the same codon where previously mutation occurs, then it is referred as first site reversion. It of two types namely exact reversion and equivalent reversion. In exact reversion type, mutation result in the same site as it occurs in the wild type where as in equivalent reversion, it occurs in different site compared to the wild type but both mutation results in the formation of same amino acid as in the wild type or result in chemically similar aminoacid.
Exact
reversion: AAA(Lys)
----------> GAA(Glu) ----------> AAA (
WILD TYPE MUTANT WILD TYPE
Equivalent reversion: UCC (Ser) ----------> UGC (Cys) ----------> AGC (Ser)
WILD TYPE MUTANT WILD TYPE
3.2.2. Second site reversion:
If reverse mutation occurs at the second site i.e. different codons, then such mutations referred as second site reversion or suppressor mutations. These suppressor mutations are further divided into two groups namely Intragenic and Extragenic suppressor mutations.
In Intragenic suppressor mutations, mutation occurs within the gene. The change occurring at the other codon suppresses the change produced by the previous mutation within the gene in this type of mutation. In Extragenic suppressor mutations, Second site mutation occurs at different genes.
4. PHENOTYPIC MUTATIONS:
Depending upon the phenotypic consequences of mutations, it might be classified as Morphological, Lethal, Conditional, Biochemical, and Loss of function and Gain of function Mutations.
Morphological Mutations:
When mutations affect the outwardly visible properties of an organism then such mutations referred as Morphological mutations. For example: Curly wings in Drosophila and dwarf peas.
Lethal Mutations:
When mutations affect the survival of the organisms then such mutations are known as lethal mutations. For example: Mutation in all Hb genes.
Conditional Mutations:
In the class of conditional mutations, a mutant allele causes a mutant phenotype in only a certain environment, called the restrictive condition, but causes a wild type phenotype in some different environment, called the permissive condition. Geneticists have studied many temperature conditional mutations. For example, certain Drosophila mutations are known as dominant heat sensitive lethal. Heterozygotes (H+/H) are wild type at 20*C (Permissive condition) but die if the temperature is raised to 30*C (the Restrictive condition).
Biochemical Mutations:
When mutations cause the loss or change of some biochemical function of the cell then such mutations are referred as Biochemical mutations. For example, one class of biochemically mutant fungi will not grow unless supplied with the nitrogenous base adenine. They are called ad mutations, whereas the wild type (Prototrophic) allele is ad+. Mutant ad alleles determine the auxotrophic, adenine-requiring phenotype.
Loss-of-function mutations:
When mutations result in the loss of function then such mutations known as Loss-of-function mutation. It is mostly found to be recessive in diploid cells. For example, mutation occurs in alpha gene of Hb.
Gain-of-function mutations:
Sometimes mutations confer some new function on the gene then such mutations referred as gain-of-function.
5. CHROMOSOMAL MUTATIONS:
Any type of change in the chromosome structure or number is referred as chromosme aberration or chromosome mutation. Chromosome mutation is the process of change that results in rearranged chromosome parts, abnormal numbers of individual chromosomes, or abnormal numbers of chromosome sets. When chromosome mutation compared with gene mutation, later is applied both to the process and to the product. Sometimes chromosome mutations can be detected by microscopic examination, sometimes by genetic analysis and sometimes by both whereas gene mutations are never detectable microscopically on a chromosome. In general, chromosome mutation is of two types namely chromosomal structural mutations and chromosomal number mutations.
5.1. CHROMOSOMAL STRUCTURAL MUTATIONS:
Following are the few types of changes, which result in chromosomal structural mutations.
5.1.1. DELETIONS:
Deletions mainly are of four types namely interstitial deletion, terminal deletion, intragenic deletion and multigenic deletion. Break at two regions result in interstitial deletion; but breakage at terminal region result in terminal deletion. They are represented in the following figure.
![]()
A small deletion within a gene, called as intragenic deletion, which result in the inactivation of gene product. When deletions result in the loss of several genes then deletions referred as multigenic deletions. Deletions are recognized genetically by reduced recombinant frequency, Pseudodominance, recessive lethality and lack of reverse mutation and cytologically by deletion loops. Deletions result in several solid tumours, which are indicated in the following figure:
5.1.2. DUPLICATIONS:
Duplications are of two types i.e., tandem and reverse duplications. When duplication occur in a continuous region then it referred as tandem duplication where duplication occurs in reverse order then duplication referred as reverse duplication. Both of them explained in the following figure:
![]()
Duplications supply additional genetic material capable of evolving new functions.
5.1.3. INVERSIONS:
Inversion is of two types namely paracentric and pericentric inversions. If inversion occurs outside the centromere then inversion referred as paracentric inversion whereas if inversion occurs in the region spanning centromere them it is called as pericentric inversion. They are diagrammatically shown below:

5.1.4. TRANSLOCATIONS:
Translocations lead to the transfer of fragments between two different chromosomes. Since parts are exchanged between two different chromosomes, it is otherwise known as reciprocal translocation. Translocations lead to the formation of different type of solid tumors in humans.
![]()
5.2. CHROMOSOMAL NUMBER MUTATIONS:
The number of chromosome is indicated by the suffix "Ploidy". There were different types of ploidies namely Euploidy, Monoploidy, Diploidy, Polyploidy and aneuploidy.
Euploid: When an organism contains normal set of chromosomes then it is known as euploid.
Monoploid: When an organism contains single set of chromosomes then it is referred as monoploid.
Diploid: When an organism contain two set of chromosomes then it is referred as diploid.
Polyploid: When an organism contain more than two set of chromosomes then it is called as Polyploid.
Aneuploid: When an organism contains chromosome number that differs from the normal chromosome number for the species by a small number of chromosomes then it is known as aneuploid.
5.2.1. MONOPLOIDY:
When organisms contain single set of chromosomes but the normal cell contain two set of chromosome then the condition referred as monoploidy. This is an chromosomal aberration because organism lacks single set of chromosome. Monoploid plants play an important role in modern approaches to plant breeding because Diploid nature is difficult to induce and select new gene mutations and study their effects. Monoploid plants are produced by tissue culture in the following steps: Pollen grains (haploid) are treated so that they will grow and are placed on agar plates containing certain plant hormones. Under these conditions, haploid embryoids will grow into monoploid plantlets. After having been moved to a medium containing different plant hormones, these plantlets will grow into mature monoploid plants with roots, stems, leaves, and flowers. To produce new plant lines, geneticists produce monoploids with favorable genotypes and then double the chromosomes to form fertile, homozygous diploids.
5.2.2. POLYPLOIDY:
There are two types of polyploids namely autopolyploids, and allopolyploids. Autopolyploids composed of multiple sets of chromosomes from within one species. Allopolyploids composed of sets from different species, which are closely related, however, the different chromosome sets are homeologous not fully homologous. Autotetraploids arise naturally by the spontaneous accidental doubling of a 2x genome to a 4x genome and Autotetraploids can be induced artificially through the use of colchicine. Autotetraploid plants are advantageous as commercial crops because in plants, the larger number of chromosome sets often leads to increased size. Cell size, fruit size, flower size and so forth, can be larger in the polyploid. Different ways of formation of tetraploids shown in the following figure:
Of the three possibilities, two bivalents and one quadrivalent possibilities form tetraploids whereas univalent-trivalent combination yields non-functional gametes.
Allopolyploid synthesized to make fertile hybrid varieties mainly. An allopolyploid fertile hybrid produced from the leaves of the cabbage and the roots of the radish. Each of these has 18 chromosomes and a viable hybrid progeny produced from seed. Most of the species found to be sterile whereas few species produce seeds. On planting, these seeds produced fertile individuals with 36 chromosomes. Allopolyploids possessing double chromosome number than their parental plant then the allopolyploids referred as amphidiploids.
In nature, allopolyploidy seems to have been a major force in speciation of plants. In the following figure three different parent species have hybridized in all possible pair combinations to form new amphidiploid species. Allopolyploids can be synthesized either by crossing related species and doubling the chromosomes of the hybrid or by asexually fusing the cells of different species.
5.2.3. ANEUPLOIDY:
Aneuploidy is the second major category of chromosome mutations in which chromosome number is abnormal. Aneuploid is an individual organism whose chromosome number differs from the wild type by part of a chromosome set. It has different conditions namely monosomic (2n-1), disomic (n+1), trisomic (2n+1) and nullisomic (2n-2).
5.2.3.1. Monosomic:
It is the condition where one chromosome less than a normal chromosome count in a species. It is generally deleterious because of two reasons. First, If the lacking chromosome present in one copy then gene balance affected and Second, if a person is recessive to certain diseases then the complementary gene containing chromosome lost then the person become diseased. Nondisjuction in mitosis or meiosis is the cause of most aneuploids. Nondisjunction is a failure of chromosome segregation process, and two chromosomes go to one pole and none to the other.
Turner Syndrome is an example for monosomic condition where one sex chromosome absent i.e. they have 44 autosomes + 1 X. Affected people have a characteristic, easily recognizable phenotype. They are sterile females, are short in stature, and often have a web of skin extending between the neck and shoulders. Although their intelligence is near normal, some of their specific cognitive functions are defective. About 1 in 5000 female births have this monosomic chromosomal complement. Monosomics for all human autosomes die in utero.
5.2.3.2. Disomic (n+1):
A disomic is an aberration of a haploid organism. In fungi, they can result from meiotic nondisjunction. Disomics in fungi can be selected from asci showing special spore abortion patterns or as meiotic progeny that must contain homologous chromosomes from both parents.
5.2.3.3. Trisomics (2n+1):
The trisomic condition also is one of chrosomal imbalance and can result in abnormality or death. However, there are many examples of viable trisomics. In other words trisomic individuals possess one extra chromosome along with their two set of chromosomes.
Klinefelter syndrome is an example for trisomics in humans. These patients are males with lanky builds who are mentally retarded and sterile. Their chromosome make up is 44 autosomes + XXY.
Down Syndrome is also another trisomic disorder but in this case trisomy occur at the 21st chromosome set. It occuring at a frequency of about 0.15% of all live births.
5.2.3.4. Nullisomics (2n-2):
It is a condition where two chromosome of a set in diploid found to be absent. This condition was normally seen in wheat plants. Although nullisomy is a lethal condition in diploids, an organism such as bread wheat, which behaves meiotically like a diploid although it is a hexaploid, can tolerate nullisomy. Their appearances differ from the normal hexaploids; furthermore, most of the nullisomics grow less vigorously.
REPAIR
MECHANISMS:
When there is damage occurs to
DNA then it should be repaired otherwise it will affect the present and
future generations. DNA, like any other molecule, can undergo a variety
of chemical reactions. Because DNA uniquely serves as a permanent copy of the
cell genome, however, changes in its structure are of much greater consequence
than are alterations in other cell components, such as RNAs or proteins.
Mutations can result from the incorporation of incorrect bases during DNA
replication. In addition, various chemical changes occur in DNA either
spontaneously or as a result of exposure to chemicals or radiation. Such
damage to DNA can block replication or transcription, and can result in a high
frequency of mutations![]() consequences
that are unacceptable from the standpoint of cell reproduction. To maintain the
integrity of their genomes, cells have therefore had to evolve mechanisms to
repair damaged DNA. Repair system generally is of four types namely direct
reversal type, Excision repair, Recombinational repair and SOS Repair.
consequences
that are unacceptable from the standpoint of cell reproduction. To maintain the
integrity of their genomes, cells have therefore had to evolve mechanisms to
repair damaged DNA. Repair system generally is of four types namely direct
reversal type, Excision repair, Recombinational repair and SOS Repair.
1. DIRECT REVERSAL OF DNA DAMAGE:
Most damage to DNA is repaired by removal of the damaged bases followed by resynthesis of the excised region. Some lesions in DNA, however, can be repaired by direct reversal of the damage, which may be a more efficient way of dealing with specific types of DNA damage that occur frequently. Only a few types of DNA damage are repaired in this way, particularly pyrimidine dimers resulting from exposure to ultraviolet (UV) light and alkylated guanine residues that have been modified by the addition of methyl or ethyl groups at the O6 position of the purine ring.
1.1. PHOTO REACTIVATION:
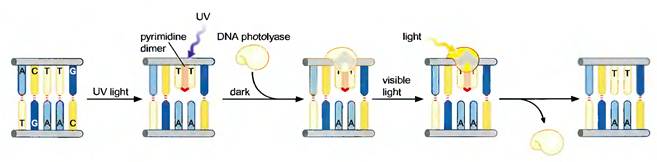
UV light is one of the major
sources of damage to DNA and is also the most thoroughly studied form of DNA
damage in terms of repair mechanisms. Its importance is illustrated by the fact
that exposure to solar UV irradiation is the cause of almost all skin cancer in
humans. The major type of damage induced by UV light is the formation of
pyrimidine dimers, in which adjacent pyrimidines on the same strand of DNA are
joined by the formation of a cyclobutane ring resulting from saturation of the
double bonds between carbons 5 and 6. The formation of such dimers distorts the
structure of the DNA chain and blocks transcription or replication past the
site of damage, so their repair is closely correlated with the ability of cells
to survive UV irradiation. One mechanism of repairing UV-induced pyrimidine
dimers is direct reversal of the dimerization reaction. The cyclobutane
pyrimidine photodimer can be repaired by a photolyase that has been found in bacteria and lower eukaryotes
but not in humans. The process is called photoreactivation because energy
derived from visible light is utilized to break the cyclobutane ring structure.
The original pyrimidine bases remain in DNA now restored to their normal state.
As might be expected from the fact that solar UV irradiation is a major source
of DNA damage for diverse cell types, the repair of pyrimidine dimers by
photoreactivation is common to a variety of prokaryotic and eukaryotic cells,
including E. coli, yeasts, and some species of plants and
animals. Curiously, however, photoreactivation is not universal; many species
(including humans) lack this mechanism of DNA repair. This enzyme cannot
operate in the dark, so other repair pathways are required to remove UV damage.
A photolyase that reverses the 6-4 photoproducts has been detected in plants
and Drosophila.
1.2. ALKYLTRANSFERASE REPAIR SYSTEM:
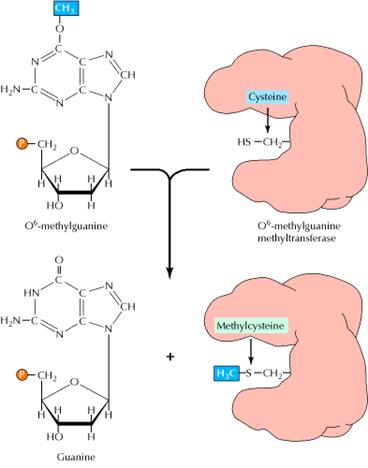
Another form of direct repair deals with damage resulting from the reaction between alkylating agents and DNA. Alkylating agents are reactive compounds that can transfer methyl or ethyl groups to a DNA base, thereby chemically modifying the base. A particularly important type of damage is methylation of the O6 position of guanine, because the product, O6-methylguanine, forms complementary base pairs with thymine instead of cytosine. This lesion can be repaired by an enzyme called O6-methylguanine methyltransferase that transfers the methyl group from O6-methylguanine to a cysteine residue in its active site. The potentially mutagenic chemical modification is thus removed, and the original guanine is restored. Enzymes that catalyze this direct repair reaction are widespread in both prokaryotes and eukaryotes, including humans.
2. EXCISION REPAIR SYSTEM:
Excision repair system includes
methods, which repairs the damage by exchanging the damaged bases or
nucleotides. Although direct repair is an efficient way of dealing with particular
types of DNA damage, excision repair is a more general means of repairing a
wide variety of chemical alterations to DNA. Consequently, the various types of
excision repair are the most important DNA repair mechanisms in both
prokaryotic and eukaryotic cells. In excision repair, the damaged DNA is
recognized and removed, either as free bases or as nucleotides. The resulting
gap is then filled in by synthesis of a new DNA strand, using the undamaged
complementary strand as a template. Three types of excision repair![]() base-excision
repair, nucleotide-excision repair, and mismatch repair
base-excision
repair, nucleotide-excision repair, and mismatch repair![]() enable
cells to cope with a variety of different kinds of DNA damage.
enable
cells to cope with a variety of different kinds of DNA damage.
2.1. BASE EXCISION REPAIR SYSTEM:
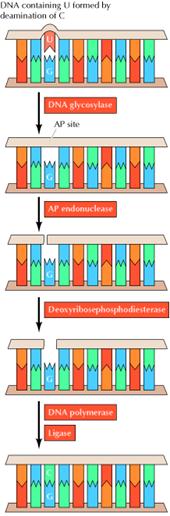
Uracil can arise in DNA by two
mechanisms: (1) Uracil (as dUTP [deoxyuridine
triphosphate]) is occasionally incorporated in place of thymine during DNA
synthesis, and (2) uracil can be formed in DNA by the deamination of cytosine.
The second mechanism is of much greater biological significance because it
alters the normal pattern of complementary base pairing and thus represents a
mutagenic event. The excision of uracil in DNA is catalyzed by DNA glycosylase,
an enzyme that cleaves the bond linking the base (uracil) to the deoxyribose of
the DNA backbone. This reaction yields free uracil and an apyrimidinic site![]() a
sugar with no base attached. DNA glycosylases also recognize and remove other
abnormal bases, including hypoxanthine formed by the deamination of adenine,
pyrimidine dimers, alkylated purines other than O6-alkylguanine, and
bases damaged by oxidation or ionizing radiation. The result of DNA glycosylase
action is the formation of an apyridiminic or
apurinic site (generally called an AP site) in DNA. Similar AP sites are formed
as the result of the spontaneous loss of purine bases, which occurs at a
significant rate under normal cellular conditions. For example, each cell in
the human body is estimated to lose several thousand purine bases daily. These
sites are repaired by AP endonuclease, which cleaves adjacent to the AP site.
The remaining deoxyribose moiety is then removed, and DNA polymerase and ligase
fill the resulting single-base gap.
a
sugar with no base attached. DNA glycosylases also recognize and remove other
abnormal bases, including hypoxanthine formed by the deamination of adenine,
pyrimidine dimers, alkylated purines other than O6-alkylguanine, and
bases damaged by oxidation or ionizing radiation. The result of DNA glycosylase
action is the formation of an apyridiminic or
apurinic site (generally called an AP site) in DNA. Similar AP sites are formed
as the result of the spontaneous loss of purine bases, which occurs at a
significant rate under normal cellular conditions. For example, each cell in
the human body is estimated to lose several thousand purine bases daily. These
sites are repaired by AP endonuclease, which cleaves adjacent to the AP site.
The remaining deoxyribose moiety is then removed, and DNA polymerase and ligase
fill the resulting single-base gap.
2.1.1. GO REPAIR SYSTEM:
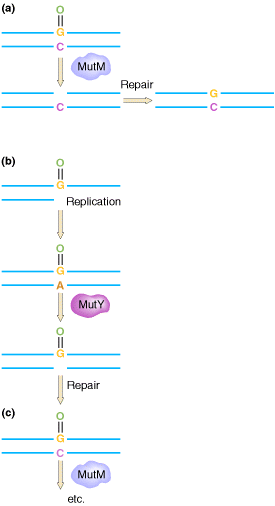
The GO REPAIR system acts in three different ways. They are as follows:
(a) 8-OxodG lesions are removed by the MutM protein, leaving an AP site that is repaired by endonucleases and repair synthesis.
(b) However, when
replicating polymerases are allowed to operate across from the lesion, they
usually add an A residue. This mispair would result in the GC ![]() TA
change, but the MutY protein removes the A, allowing repair of the resulting AP
site.
TA
change, but the MutY protein removes the A, allowing repair of the resulting AP
site.
(c) When repair polymerases operate across from the 8-oxodG lesion, they preferentially restore a C across from the lesion, allowing the MutM protein another opportunity to remove the lesion.
The mutT product prevents incorporation of GO across from A. The human counterparts of the mutT, mutY, and mutM gene products have been detected.
2.2. NUCLEOTIDE EXCISION REPAIR SYSTEM:
2.2.1. UvrABC Excinuclease system:
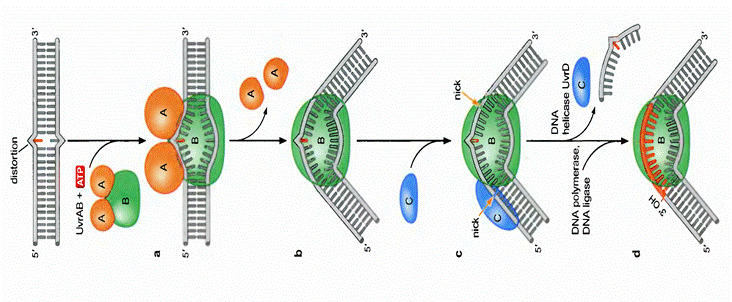
In E. coli,
nucleotide-excision repair is catalyzed by the products of three genes (uvrA, B, and C) that were identified
because mutations at these loci result in extreme sensitivity to UV light. The
protein UvrA recognizes damaged DNA and recruits UvrB and UvrC to the site of the
lesion. UvrB and UvrC then
cleave on the 3![]() and 5
and 5![]() sides of the damaged site, respectively, thus excising an oligonucleotide
consisting of 12 or 13 bases. The UvrABC complex is
frequently called an excinuclease,
a name that reflects its ability to directly excise an oligonucleotide.
The action of a helicase is then required to remove the damage-containing
oligonucleotide from the double-stranded DNA molecule, and the resulting gap is
filled by DNA polymerase I and sealed by ligase. The human excinuclease
is considerably more complex than its bacterial counterpart and includes at
least 17 proteins. However, the basic steps are the same as those in E.
coli.
sides of the damaged site, respectively, thus excising an oligonucleotide
consisting of 12 or 13 bases. The UvrABC complex is
frequently called an excinuclease,
a name that reflects its ability to directly excise an oligonucleotide.
The action of a helicase is then required to remove the damage-containing
oligonucleotide from the double-stranded DNA molecule, and the resulting gap is
filled by DNA polymerase I and sealed by ligase. The human excinuclease
is considerably more complex than its bacterial counterpart and includes at
least 17 proteins. However, the basic steps are the same as those in E.
coli.
2.2.2. EXCISION REPAIR SYSTEM IN EUKARYOTES:
In mammalian cells, the XPA
protein (and possibly also XPC) initiates repair by recognizing damaged DNA and
forming complexes with other proteins involved in the repair process. These
include the XPB and XPD proteins, which act as helicases that unwind the
damaged DNA. In addition, the binding of XPA to damaged DNA leads to the
recruitment of XPF (as a heterodimer with ERCC1) and XPG to the repair complex.
XPF/ERCC1 and XPG are endonucleases, which cleave DNA on the 5![]() and 3
and 3![]() sides of the damaged site, respectively. This cleavage excises an
oligonucleotide consisting of approximately 30 bases. The resulting gap then
appears to be filled in by DNA polymerase d or e (in
association with RFC and PCNA) and sealed by ligase.
sides of the damaged site, respectively. This cleavage excises an
oligonucleotide consisting of approximately 30 bases. The resulting gap then
appears to be filled in by DNA polymerase d or e (in
association with RFC and PCNA) and sealed by ligase.
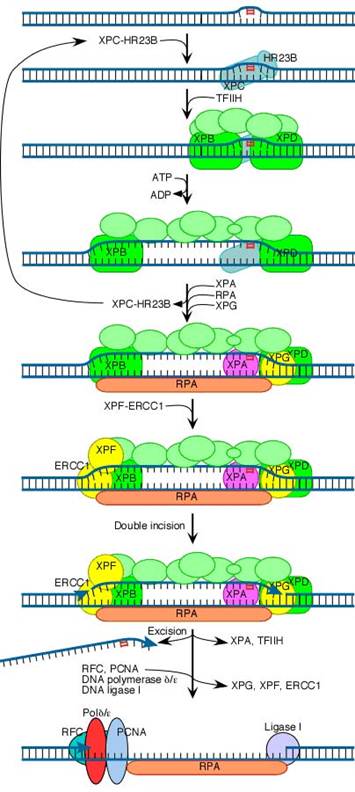
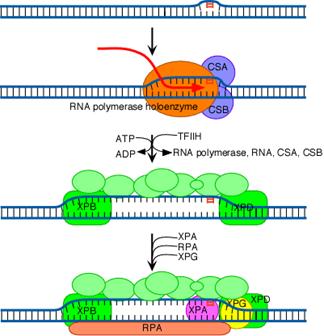
The initial steps depend on whether the damage is in the actively transcribed strand of a gene or elsewhere in the genome. If the damage is not in the actively transcribed strand of a gene, then the damage is recognized and bound by a heterodimer consisting of the XPC and HR23B proteins. The binding of XPC and HR23B initiates the process of "global genome repair" (GGR), which simply means repair anywhere in the genome.
The XPC/HR23B dimer appears to recognize damaged DNA based on the extent of distortion of the normal helical DNA structure caused by the damage. In the process of binding to the damaged region, XPC/HR23B is thought to further increase the extent of structural distortion, as illustrated in this diagram.
The increased distortion produced by XPC/HR23B permits the entry and binding of the general transcription factor TFIIH, whose 10 subunits are colored in various shades of green in the above diagram. Two of these subunits (XPB and XPD) are helicases, which bind to the damaged strand and use the energy of ATP to unwind a stretch of 20-30 nucleotides including the damaged site.
Three additional proteins then bind to and stabilize the open complex. The precise role of XPA is unclear, but evidence suggests that it checks to confirm that damage is present in the opened region and assists in stabilizing the open complex. RPA is the major eukaryotic single-stranded-DNA-binding protein. It binds to and protects both of the separated strands in the open complex. For clarity in the diagram, it is shown binding only to the bottom strand. XPG is a structure-specific nuclease.
Concomitant with the binding of XPA, RPA and XPG, XPC and HR23B are released. These two proteins are then free to recycle to other damaged sites where the repair process has not yet been initiated.
The next step in the repair process, for both GGR and TC-NER, is recruitment of another structure-specific endonuclease, the XPF-ERCC1 heterodimer:
Both XPG and XPF-ERCC1 are specific for junctions between single- and double-stranded DNA. XPG, which is closely related to the FEN-1 nuclease that participates in base excision repair, cuts on the 3' side of such a junction, while ERCC1/XPF (a heterodimeric protein complex) cuts on the 5' side.
The cut made by XPG is 2-8 nucleotides from the lesion, and the cut made by ERCC1/XPF is 15-24 nucleotides away. These distances are paired with each other (probably as a consequence of the structure of the multiprotein complex) in such a way that the damage-containing oligonucleotide between the cuts averages 27 nucleotides (range 24-32 nucleotides).
The damage-containing oligonucleotide is displaced concomitant with the binding of replicative gap-repair proteins (RFC, PCNA, DNA polymerase delta or epsilon), with the displacement of TFIIH, XPA, XPG, and XPF-ERCC1, and with new DNA synthesis that fills the gap. The final nick is sealed by DNA ligase I.
2.2.3. REPAIR COUPLING WITH TRANSCRIPTION:
An intriguing feature of nucleotide-excision repair is its relationship to transcription. A connection between transcription and repair was first suggested by experiments showing that transcribed strands of DNA are repaired more rapidly than nontranscribed strands in both E. coli and mammalian cells. Since DNA damage blocks transcription, this transcription-repair coupling is thought to be advantageous by allowing the cell to preferentially repair damage to actively expressed genes.
2.2.3.1. COUPLING IN PROKARYOTES:
In E. coli, the mechanism of transcription-repair coupling involves recognition of RNA polymerase stalled at a lesion in the DNA strand being transcribed. The stalled RNA polymerase is recognized by a protein called transcription-repair coupling factor, which displaces RNA polymerase and recruits the UvrABC excinuclease to the site of damage which intern removes the error and the transcription continued.
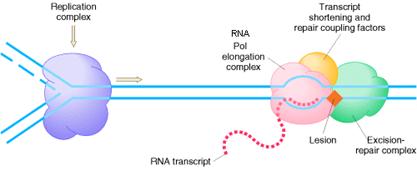
2.2.3.2. COUPLING IN EUKARYOTES:
In Cockayne’s syndrome, two proteins found to be inactive namely CSA and CSB and they are also unable to carry out transcription blocked DNA repair. When transcription stahled at the thymidine dimer damaged site, CSA and CSB bound to stahled RNA polymerase. This then recruits the binding of TFII H factor to the site but RNA, RNA polymerase, CSA and CSB are released. The remaining reactions are same as in Nucleotide Excision repair system of eukaryotes. Thus CSA and CSB were similar to XP C and HR23B in function.
2.3. MISMATCH REPAIR SYSTEM:
Some repair pathways are capable of recognizing errors even after DNA replication has already occurred. One such system, termed the mismatch repair system, can detect mismatches that occur in DNA replication. It occurs mainly in three steps namely
1. Recognize mismatched base pairs.
2. Determine, which base in the mismatch, is the
incorrect one.
3. Excise the incorrect base and carry out
repair synthesis.
The second point is the crucial property of such a system. Unless it is
capable of discriminating between the correct and the incorrect bases, the
mismatch repair system could not determine which base to excise. If, for
example, a G![]() T
mismatch occurs as a replication error, how can the system determine whether G
or T is incorrect? Both are normal bases in DNA. But replication errors produce
mismatches on the newly synthesized strand, so it is the base on this strand
that must be recognized and excised. To distinguish the old, template strand
from the newly synthesized strand, the mismatch repair system in bacteria takes
advantage of the normal delay in the postreplication methylation of the sequence (GATC).
The methylating enzyme is adenine methylase, which creates 6-methyladenine on each
strand. However, it takes the adenine methylase several minutes to recognize
and modify the newly synthesized GATC stretches. During that interval, the
mismatch repair system can operate because it can now distinguish the old
strand from the new one by the methylation pattern. Methylating the 6-position
of adenine does not affect base pairing, and it provides a convenient tag that
can be detected by other enzyme systems. The old strand is methylated at
GATC sequences right after replication. When the mismatched site has been
identified, the mismatch repair system corrects the error.
T
mismatch occurs as a replication error, how can the system determine whether G
or T is incorrect? Both are normal bases in DNA. But replication errors produce
mismatches on the newly synthesized strand, so it is the base on this strand
that must be recognized and excised. To distinguish the old, template strand
from the newly synthesized strand, the mismatch repair system in bacteria takes
advantage of the normal delay in the postreplication methylation of the sequence (GATC).
The methylating enzyme is adenine methylase, which creates 6-methyladenine on each
strand. However, it takes the adenine methylase several minutes to recognize
and modify the newly synthesized GATC stretches. During that interval, the
mismatch repair system can operate because it can now distinguish the old
strand from the new one by the methylation pattern. Methylating the 6-position
of adenine does not affect base pairing, and it provides a convenient tag that
can be detected by other enzyme systems. The old strand is methylated at
GATC sequences right after replication. When the mismatched site has been
identified, the mismatch repair system corrects the error.
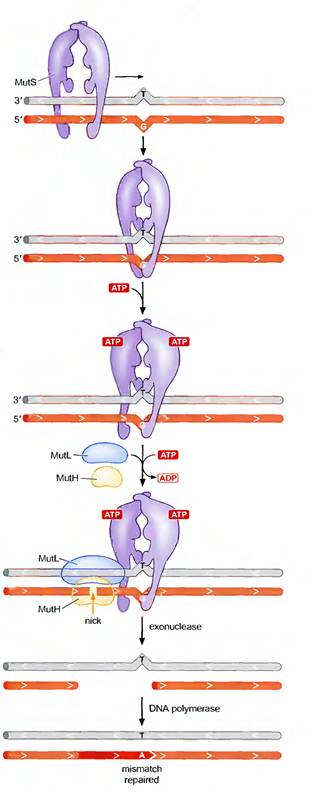
2.3.1. MISMATCH REPAIR IN E.COLI:
In E. coli, the ability of the mismatch repair system to distinguish between parental DNA and newly synthesized DNA is based on the fact that DNA of this bacterium is modified by the methylation of adenine residues within the sequence GATC to form 6-methyladenine. Since methylation occurs after replication, newly synthesized DNA strands are not methylated and thus can be specifically recognized by the mismatch repair enzymes. Mismatch repair is initiated by the protein MutS, which recognizes the mismatch and forms a complex with two other proteins called MutL and MutH. The MutH endonuclease then cleaves the unmethylated DNA strand at a GATC sequence. MutL and MutS then act together with an exonuclease and a helicase to excise the DNA between the strand break and the mismatch, with the resulting gap being filled by DNA polymerase and ligase.
2.3.2. MISMATCH REPAIR SYSTEM
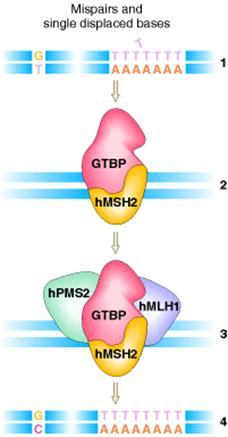
IN HUMANS:
The mismatch repair system has
also been characterized in humans. Two of the proteins, hMSH2 and hMLH1, were
very similar to their bacterial counterparts, MutS
and MutL, respectively. The hMSH2 protein, together
with the G![]() T-binding
protein (GTBP), binds to the mismatches and then recruits the other components
of the system, hPMS2 and hMLH1, to effect repair of the mismatch.
T-binding
protein (GTBP), binds to the mismatches and then recruits the other components
of the system, hPMS2 and hMLH1, to effect repair of the mismatch.
3.
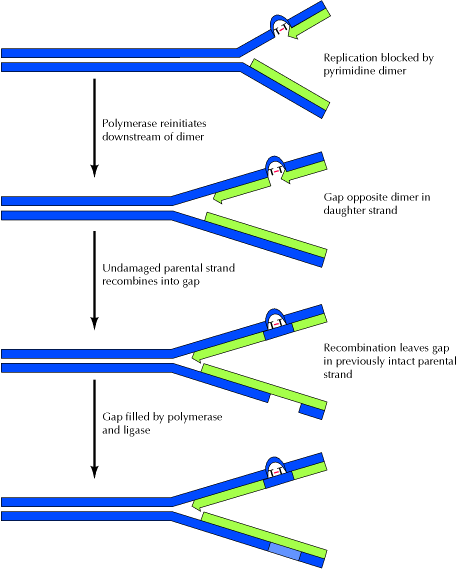
RECOMBINATION REPAIR:
The presence of a thymine dimer blocks replication, but DNA polymerase can bypass the lesion and reinitiate replication at a new site downstream of the dimer, which is referred as postdimer initiation. The result is a gap opposite the dimer in the newly synthesized DNA strand. In recombinational repair, this gap is filled by recombination with the undamaged parental strand. Although this leaves a gap in the previously intact parental strand, the gap can be filled by the actions of polymerase and ligase, using the intact daughter strand as a template.
Two intact DNA molecules are thus formed, and the remaining thymine dimer eventually can be removed by excision repair. It is otherwise known as daughter strand gap or sister strand gap repair system because only the gaps formed opposite to dimers, rather than the dimers themselves, are repaired. Since recombination repair occurs after DNA replication, in contrast with excision repair, it has been called as postreplicational repair. After recombination repair clears replication, the damage in template DNA repaired by Nucleotide Excision Repair (NER) system.
4.
SOS REPAIR:
This is an example for the
repair system, which found to be an error prone repair system. It is an
error prone process because the strands contain incorrect base eventhough intact DNA strands is formed. This repair
system otherwise known as by pass repair system because it allows DNA chain
growth across damaged segments at the cost of fidelity of replication.
The name SOS means Save Our Soul. This is because, the principle involved
is that survival with mutations is better than no survival at all.
DNA polymerase III stops at a noncoding lesion, such as the T![]() C
photodimer, generating single-stranded regions that
attract the Ssb protein and RecA,
which forms filaments. The presence of RecA filaments
helps to signal the cell to synthesize UmuD, which is
cleaved by RecA to yield UmuD
C
photodimer, generating single-stranded regions that
attract the Ssb protein and RecA,
which forms filaments. The presence of RecA filaments
helps to signal the cell to synthesize UmuD, which is
cleaved by RecA to yield UmuD![]() .
The UmuC is recruited to form a complex with UmuD
.
The UmuC is recruited to form a complex with UmuD![]() that permits DNA polymerization to proceed past the blocking lesion.
Since DNA synthesis occur even across the thymine dimer region (damaged
region), synthesis of DNA referred as transdimer
synthesis.
that permits DNA polymerization to proceed past the blocking lesion.
Since DNA synthesis occur even across the thymine dimer region (damaged
region), synthesis of DNA referred as transdimer
synthesis.
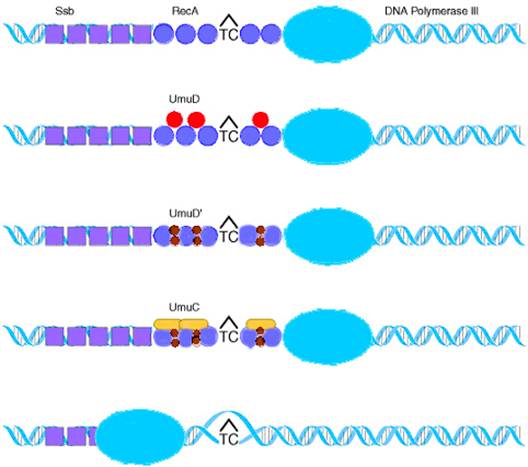
The survival of an ultraviolet-irradiated lamda virus is higher on an irradiated host than on an unirradiated host when the damage is repaired by SOS repair. Then the phenomenon is called as UV-reactivation or W reactivation. This is possible only in rec+ species only. This repair mechanism was also known as transleison DNA synthesis mechanism because it synthesis DNA in the damaged DNA.
PREVENTION OF DNA ERROR:
Some enzymatic systems neutralize potentially damaging compounds before they even react with DNA. One example of such a system is the detoxification of superoxide radicals produced during oxidative damage to DNA: the enzyme superoxide dismutase catalyzes the conversion of the superoxide radicals into hydrogen peroxide, and the enzyme catalase, in turn, converts the hydrogen peroxide into water. Another error-prevention pathway depends on the protein product of the mutT gene: this enzyme prevents the incorporation of 8-oxodG, which arises by oxidation of dGTP, into DNA by hydrolyzing the triphosphate of 8-oxodG back to the monophosphate.
SITE DIRECTED MUTAGENESIS:
Site-directed mutagenesis (SDM) is a molecular biology technique in which a mutation is created at a defined site in a DNA molecule, usually a circular molecule known as a plasmid. In general, site-directed mutagenesis requires that the wild type gene sequence be known.
This technique is also known as site-specific mutagenesis or oligonucleotide-directed mutagenesis.
By using this method, one can create mutations at any specific site in a gene whose wild-type sequence is already known. This known sequence is used to chemically synthesize short DNA segments called oligonucleotides. Through single-strand hybridization, these oligonucleotides can be directed to any chosen site in the gene. In one approach, the gene of interest is inserted into a single-stranded phage vector, such as the phage M13. A synthetic oligonucleotide containing the desired mutation is designed. This oligonucleotide is allowed to hybridize to the mutant site by complementary base pairing. The oligonucleotide serves as a primer for the in vitro synthesis of the complementary strand of the M13 vector.
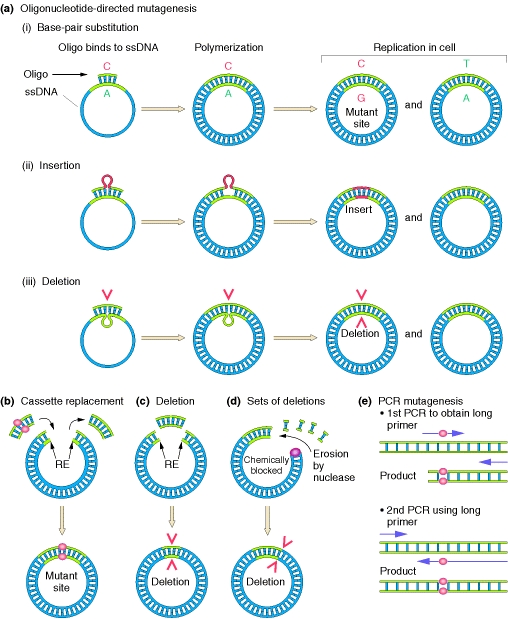
Any desired specific base change can be programmed into the sequence of the synthetic primer. Although there will be a mispaired base when the synthetic oligonucleotide hybridizes with the complementary sequence on the M13 vector, a few mismatched bases can be tolerated when hybridization takes place at a low temperature and a high salt concentration. After DNA synthesis has been mediated by DNA polymerase in vitro, the M13 DNA is allowed to replicate in E. coli, in which case many of the resulting phages will be the desired mutant. The synthetic oligonucleotide can be used as a labeled probe to distinguish wild-type from mutant phages. Although, at low temperature, the mismatched base will not prevent the primer from hybridizing with both types of phage, at high temperature, the primer will hybridize only with the mutant phage. Oligonucleotides with deletions or insertions will direct comparable mutations in the resident gene. The site-directed method can also be used on genes cloned in double-stranded vectors if the DNA is first denatured.
Knowledge of restriction sites is also useful in modifying a cloned gene. For example, a small deletion can be made by removing the fragment liberated by cutting at two restriction sites. With the use of a similar double cut, a fragment, or “cassette,” can be inserted at a single restriction cut to create duplication or other modification. Another approach is to enzymatically erode a cut end created by a restriction enzyme, to create deletions of various lengths.
The polymerase chain reaction (PCR) also can be used. A primer containing a mutation is used in a first round of PCR. The product of the first round is used as a primer for a second round of PCR, whose product is the mutant gene.
Recombination
Recombination is the production of new DNA molecule(s) from two parental DNA molecules or different segments of the same DNA molecule. Transposition is a highly specialized form of recombination in which a segment of DNA moves from one location to another, either on the same chromosome or a different chromosome.
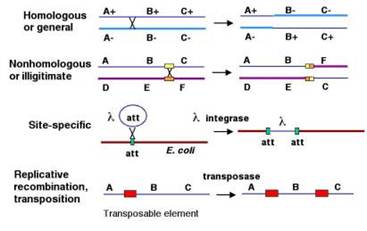
At least four types of naturally occurring recombination have been identified in living organisms. General or homologous recombination occurs between DNA molecules of very similar sequence, such as homologous chromosomes in diploid organisms. General recombination can occur throughout the genome of diploid organisms, using one or a small number of common enzymatic pathways. Illegitimate or nonhomologous recombination occurs in regions where no large-scale sequence similarity is apparent, e.g. translocations between different chromosomes or deletions that remove several genes along a chromosome. However, when the DNA sequence at the breakpoints for these events is analyzed, short regions of sequence similarity are found in some cases. For instance, recombination between two similar genes that are several million bp apart can lead to deletion of the intervening genes in somatic cells. Site-specific recombination occurs between particular short sequences (about 12 to 24 bp) present on otherwise dissimilar parental molecules. Site-specific recombination requires a special enzymatic machinery, basically one enzyme or enzyme system for each particular site. Good examples are the systems for integration of some bacteriophage, such as , into a bacterial chromosome and the rearrangement of immunoglobulin genes in vertebrate animals. The third type is replicative recombination, which generates a new copy of a segment of DNA. Many transposable elements use a process of replicative recombination to generate a new copy of the transposable element at a new location.
General
Recombination
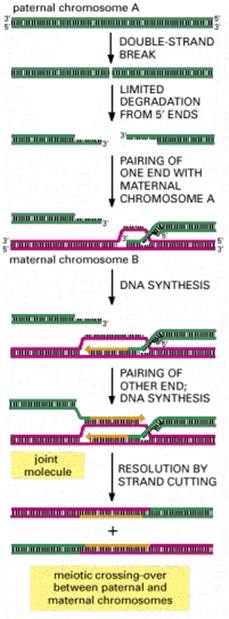
General recombination is an integral part of the complex process of meiosis in sexually reproducing organisms. It results in a crossing over between pairs of genes along a chromosome, which are revealed in appropriate matings. The chiasmata that link homologous chromosomes during meiosis are the likely sites of the crossovers that result in recombination. General recombination also occurs in nonsexual organisms when two copies of a chromosome or chromosomal segment are present. Recombination during F-factor mediated conjugal transfer of parts of chromosomes in E. coli. Recombination between two phages during a mixed infection of bacteria is another example. Also, the retrieval system for post-replicative repair involves general recombination.
General recombination can appear to result in either an equal or an unequal exchange of genetic information. Equal exchange is referred to as reciprocal recombination. In this example, two homologous chromosomes are distinguished by having wild type alleles on one chromosome (A+, B+ and C+) and mutant alleles on the other (A-, B- and C-). Homologous recombination between genes A and B exchanges the segment of one chromosome containing the wild type alleles of genes B and C (B+ and C+) for the segment containing the mutant alleles (B- and C-) on the homologous chromosome. This process resulting in new DNA molecules that carry genetic information derived from both parental DNA molecules is called reciprocal recombination. The number of alleles for each gene remains the same in the products of this recombination, only their arrangement has changed. General recombination can also result in a one-way transfer of genetic information, resulting in an allele of a gene on one chromosome being changed to the allele on the homologous chromosome. This is called gene conversion.
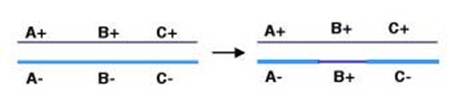
Holliday
model
One of the first plausible models to account for the preceding observations was formulated by Robin Holliday. The key features of the Holliday model are the formation of heteroduplex DNA; the creation of a cross bridge; its migration along the two heteroduplex strands, termed branch migration; the occurrence of mismatch repair; and the subsequent resolution, or splicing, of the intermediate structure to yield different types of recombinant molecules.
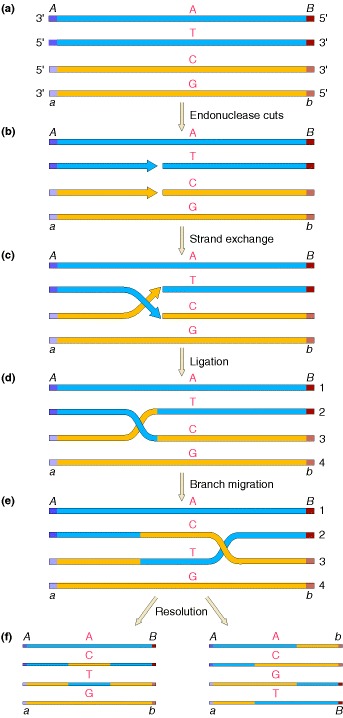
two homologous double helices are aligned, although note that they have been rotated so that the bottom strand of the first helix has the same polarity as the top strand of the second helix (5′ → 3′ in this case). Then a nuclease cleaves the two strands that have the same polarity. The free ends leave their original complementary strands and undergo hydrogen bonding with the complementary strands in the homologous double helix. Ligation produces the crossed structure. This partially heteroduplex double helix is a crucial intermediate in recombination, and has been termed the Holliday structure. The Holliday structure creates a cross bridge, or branch, that can move, or migrate, along the heteroduplex. This phenomenon of branch migration is a distinctive property of the Holliday structure. he Holliday structure can be resolved by cutting and ligating either the two originally exchanged strands or the originally unexchanged strands. The former generates a pair of duplexes that are parental, except for a stretch in the middle containing one strand from each parent. If the two parents had different alleles in this stretch, as indicated here, then the DNA will be heteroduplex. The latter resolution step generates two duplexes that are recombinant, with a stretch of heteroduplex DNA. The Holliday model also postulated that the heteroduplex DNA mismatches can be repaired by an enzymatic correction system that recognizes mismatches and excises the mismatched base from one of the two strands, filling in the excised base with the correct complementary base. The resulting molecules will carry either the wild-type or the mutant allele, depending on which allele is excised.
A Holliday junction is a branched nucleic acid structure that contains four double-stranded arms joined together. These arms may adopt one of several conformations depending on buffer salt concentrations and the sequence of nucleobases closest to the junction. The structure is named after the molecular biologist Robin Holliday, who proposed its existence in 1964.
Meselson-Radding
model
As the data from tetrad analyses accumulated, it became clear that the Holliday model could not explain everything. For instance, the two mismatches resulting from the two heteroduplexes and should be manifested in the progeny from a cross, yielding aberrant 4:4 tetrads. Yet, tetrad analyses in yeast and other organisms showed that, whereas 6:2 tetrads were frequent among gene-conversion events, aberrant 4:4 tetrads were very rare. It seemed as if gene conversion and the formation of heteroduplex DNA occurred primarily in only one chromatid. The model proposed by Meselson and Radding generates the Holliday structure with one single-strand cut in only one chromosome, in contrast with the Holliday model, in which a nick is made in one strand in each of the two homologous chromatids. This single-strand cut is followed by DNA synthesis. After the nick, the displaced single strand invades the second duplex, generating a loop, which is excised. After ligation to produce a Holliday structure, followed by branch migration, a heteroduplex is generated in each chromosome. Resolution of this intermediate occurs exactly as depicted in Figure. Note the lack of symmetry in the heteroduplex DNA at resolution in Figure f (left), compared with Figures of holiday model. Thus, in the Meselson-Radding model (left side), the bottom chromatid duplex has a heteroduplex region, instead of the two chromatids having heteroduplex regions, as in the Holliday model. However, branch migration and isomerization can generate a structure that has heteroduplex regions on both duplexes, which is required to explain aberrant 4:4 ratios.
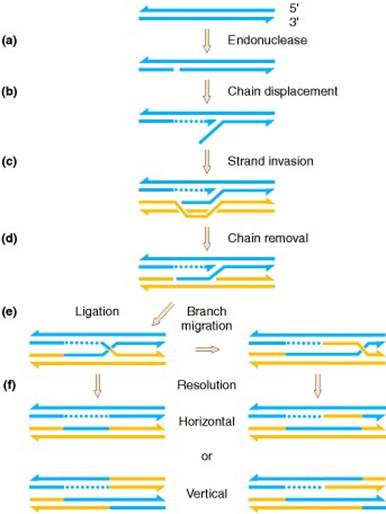
a) A duplex is cut on one chain. (b) DNA polymerase displaces one chain. (c) The resulting single chain displaces its counterpart in the homolog. (d) This displaced chain is enzymatically digested. (e) Ligation completes the formation of a Holliday junction, which is genetically asymmetric in that only one of the two duplexes has a region of potentially heteroduplex DNA. If the junction migrates, heteroduplex DNA can arise on both duplexes. (f) Resolution of the junction occurs as in the Holliday model.
Models
for general recombinations
There are four models proposed for general recombinations. They are DSBR, SDSA, SSA and BIR pathways.
Double
strand break repair model (DSBR) / Pathway
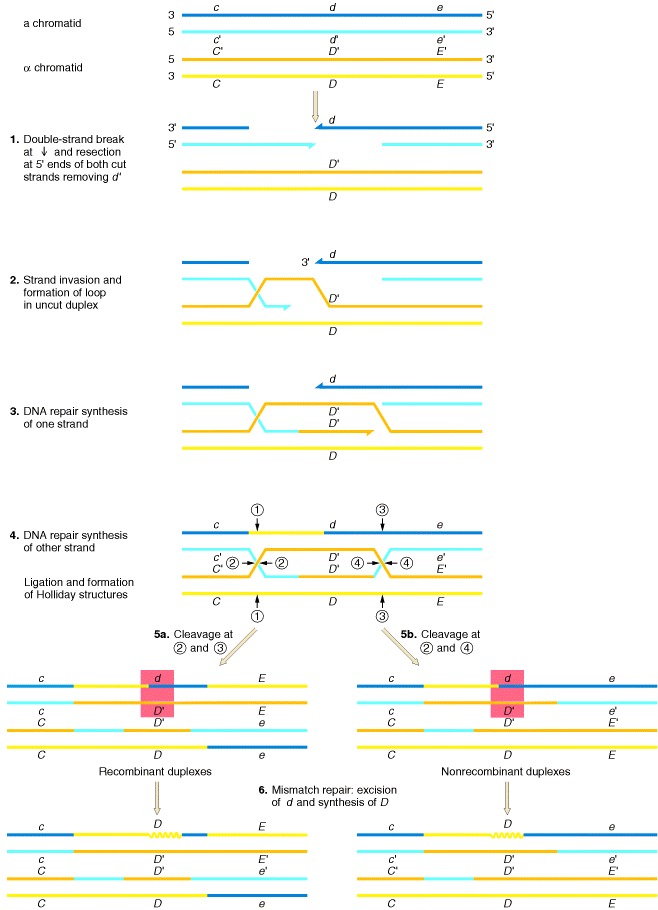
In the Holliday and Meselson-Radding models for genetic recombination, the initiation events for recombination are single-strand nicks that result in the generation of heteroduplex DNA. However, the finding that yeast transformation is stimulated 1000-fold when a double-strand break is introduced into a circular donor plasmid provided the impetus for an additional model, the double-strand-break model shown in Figure. Originally formulated by Jack Szostak, Terry Orr-Weaver, and Rodney Rothstein, this model invokes double-strand breaks to initiate recombination. The breaks are enlarged to gaps, and the repair of the double-stranded gap results in gene conversion. The key features of this model are diagrammed in the steps in Figure: (1) a double-strand break, followed by digestion of the 5′ end of both cut sites; (2) the invasion by a remaining 3′ tail of the uncut other duplex; (3) the repair synthesis of one strand; (4) the repair synthesis of the other strand, and ligation to form two Holliday junctions; (5) resolution in one of two ways, one of which generates a reciprocal crossover; and (6) mismatch repair correction to yield gene conversion.
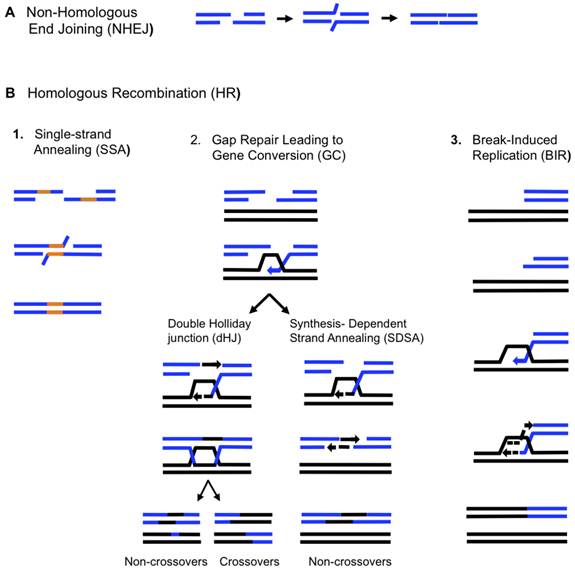
SDSA
Pathway
Homologous recombination via the SDSA pathway occurs in cells that divide through mitosis and meiosis and results in non-crossover products. In this model, the invading 3' strand is extended along the recipient DNA duplex by a DNA polymerase, and is released as the Holliday junction between the donor and recipient DNA molecules slides in a process called branch migration. The newly synthesized 3' end of the invading strand is then able to anneal to the other 3' overhang in the damaged chromosome through complementary base pairing. After the strands anneal, a small flap of DNA can sometimes remain. Any such flaps are removed, and the SDSA pathway finishes with the resealing, also known as ligation, of any remaining single-stranded gaps.
During mitosis, the major homologous recombination pathway for repairing DNA double-strand breaks appears to be the SDSA pathway (rather than the DSBR pathway). The SDSA pathway produces non-crossover recombinants. During meiosis non-crossover recombinants also occur frequently and these appear to arise mainly by the SDSA pathway as well. Non-crossover recombination events occurring during meiosis likely reflect instances of repair of DNA double-strand damages or other types of DNA damages.
SSA
Pathway
The single-strand annealing (SSA) pathway of homologous recombination repairs double-strand breaks between two repeat sequences. The SSA pathway is unique in that it does not require a separate similar or identical molecule of DNA, like the DSBR or SDSA pathways of homologous recombination. Instead, the SSA pathway only requires a single DNA duplex, and uses the repeat sequences as the identical sequences that homologous recombination needs for repair. The pathway is relatively simple in concept: after two strands of the same DNA duplex are cut back around the site of the double-strand break, the two resulting 3' overhangs then align and anneal to each other, restoring the DNA as a continuous duplex.
As DNA around the double-strand break is cut back, the single-stranded 3' overhangs being produced are coated with the RPA protein, which prevents the 3' overhangs from sticking to themselves. A protein called Rad52 then binds each of the repeat sequences on either side of the break, and aligns them to enable the two complementary repeat sequences to anneal. After annealing is complete, leftover non-homologous flaps of the 3' overhangs are cut away by a set of nucleases, known as Rad1/Rad10, which are brought to the flaps by the Saw1 and Slx4 proteins. New DNA synthesis fills in any gaps, and ligation restores the DNA duplex as two continuous strands. The DNA sequence between the repeats is always lost, as is one of the two repeats. The SSA pathway is considered mutagenic since it results in such deletions of genetic material.
BIR
Pathway
During DNA replication, double-strand breaks can sometimes be encountered at replication forks as DNA helicase unzips the template strand. These defects are repaired in the break-induced replication (BIR) pathway of homologous recombination. The precise molecular mechanisms of the BIR pathway remain unclear. Three proposed mechanisms have strand invasion as an initial step, but they differ in how they model the migration of the D-loop and later phases of recombination.
The BIR pathway can also help to maintain the length of telomeres (regions of DNA at the end of eukaryotic chromosomes) in the absence of (or in cooperation with) telomerase. Without working copies of the telomerase enzyme, telomeres typically shorten with each cycle of mitosis, which eventually blocks cell division and leads to senescence. In budding yeast cells where telomerase has been inactivated through mutations, two types of "survivor" cells have been observed to avoid senescence longer than expected by elongating their telomeres through BIR pathways.
Maintaining telomere length is critical for cell immortalization, a key feature of cancer. Most cancers maintain telomeres by upregulating telomerase. However, in several types of human cancer, a BIR-like pathway helps to sustain some tumors by acting as an alternative mechanism of telomere maintenance. This fact has led scientists to investigate whether such recombination-based mechanisms of telomere maintenance could thwart anti-cancer drugs like telomerase inhibitors.
Recombination
in E.Coli (Bacteria)
Homologous recombination is a major DNA repair process in bacteria. It is also important for producing genetic diversity in bacterial populations, although the process differs substantially from meiotic recombination, which repairs DNA damages and brings about diversity in eukaryotic genomes. Homologous recombination has been most studied and is best understood for Escherichia coli. Double-strand DNA breaks in bacteria are repaired by the RecBCD pathway of homologous recombination. Breaks that occur on only one of the two DNA strands, known as single-strand gaps, are thought to be repaired by the RecF pathway. Both the RecBCD and RecF pathways include a series of reactions known as branch migration, in which single DNA strands are exchanged between two intercrossed molecules of duplex DNA, and resolution, in which those two intercrossed molecules of DNA are cut apart and restored to their normal double-stranded state.
RecBCD Pathway
The RecBCD pathway is the main recombination pathway used in many bacteria to repair double-strand breaks in DNA, and the proteins are found in a broad array of bacteria. These double-strand breaks can be caused by UV light and other radiation, as well as chemical mutagens. Double-strand breaks may also arise by DNA replication through a single-strand nick or gap. Such a situation causes what is known as a collapsed replication fork and is fixed by several pathways of homologous recombination including the RecBCD pathway.
In this pathway, a three-subunit enzyme complex called RecBCD initiates recombination by binding to a blunt or nearly blunt end of a break in double-strand DNA. After RecBCD binds the DNA end, the RecB and RecD subunits begin unzipping the DNA duplex through helicase activity. The RecB subunit also has a nuclease domain, which cuts the single strand of DNA that emerges from the unzipping process. This unzipping continues until RecBCD encounters a specific nucleotide sequence (5'-GCTGGTGG-3') known as a Chi site.
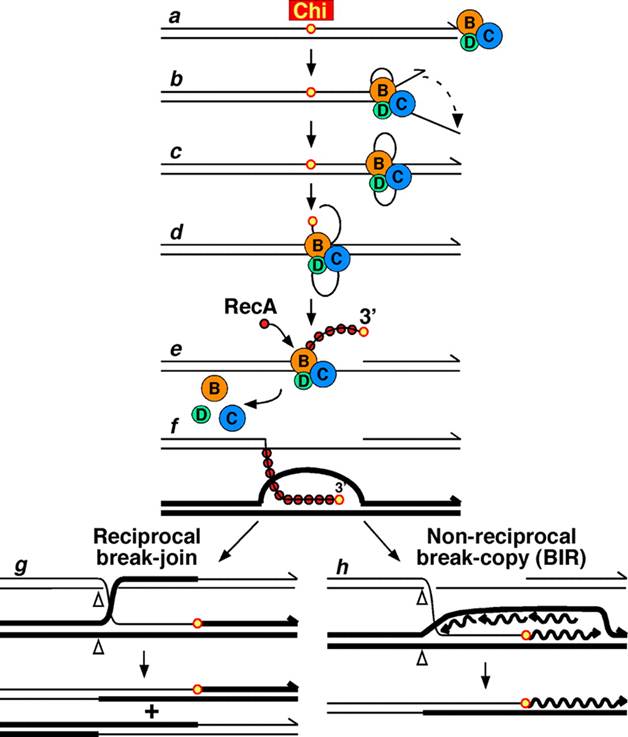
Upon encountering a Chi site, the activity of the RecBCD enzyme changes drastically. DNA unwinding pauses for a few seconds and then resumes at roughly half the initial speed. This is likely because the slower RecB helicase unwinds the DNA after Chi, rather than the faster RecD helicase, which unwinds the DNA before Chi. Recognition of the Chi site also changes the RecBCD enzyme so that it cuts the DNA strand with Chi and begins loading multiple RecA proteins onto the single-stranded DNA with the newly generated 3' end. The resulting RecA-coated nucleoprotein filament then searches out similar sequences of DNA on a homologous chromosome. The search process induces stretching of the DNA duplex, which enhances homology recognition (a mechanism termed conformational proofreading). Upon finding such a sequence, the single-stranded nucleoprotein filament moves into the homologous recipient DNA duplex in a process called strand invasion. The invading 3' overhang causes one of the strands of the recipient DNA duplex to be displaced, to form a D-loop. If the D-loop is cut, another swapping of strands forms a cross-shaped structure called a Holliday junction. Resolution of the Holliday junction by some combination of RuvABC or RecG can produce two recombinant DNA molecules with reciprocal genetic types, if the two interacting DNA molecules differ genetically. Alternatively, the invading 3’ end near Chi can prime DNA synthesis and form a replication fork. This type of resolution produces only one type of recombinant (non-reciprocal).
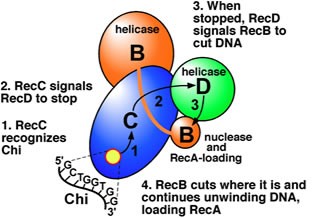
Molecular model for the RecBCD pathway of recombination. This model is based on reactions of DNA and RecBCD with ATP in excess over Mg2+ ions. Step 1: RecBCD binds to a double-stranded DNA end. Step 2: RecBCD unwinds DNA. RecD is a fast helicase on the 5’-ended strand, and RecB is a slower helicase on the 3’-ended strand (that with an arrowhead) [ref 46 in current Wiki version]. This produces two single-stranded (ss) DNA tails and one ss loop. The loop and tails enlarge as RecBCD moves along the DNA. Step 3: The two tails anneal to produce a second ss DNA loop, and both loops move and grow. Step 4: Upon reaching the Chi hotspot sequence (5’ GCTGGTGG 3’; red dot) RecBCD nicks the 3’-ended strand. Further unwinding produces a long 3’-ended ss tail with Chi near its end. Step 5: RecBCD loads RecA protein onto the Chi tail. At some undetermined point, the RecBCD subunits disassemble. Step 6: The RecA-ssDNA complex invades an intact homologous duplex DNA to produce a D-loop, which can be resolved into intact, recombinant DNA in two ways. Step 7: The D-loop is cut and anneals with the gap in the first DNA to produce a Holliday junction. Resolution of the Holliday junction (cutting, swapping of strands, and ligation) at the open arrowheads by some combination of RuvABC and RecG produces two recombinants of reciprocal type. Step 8: The 3’ end of the Chi tail primes DNA synthesis, from which a replication fork can be generated. Resolution of the fork at the open arrowheads produces one recombinant (non-reciprocal) DNA, one parental-type DNA, and one DNA fragment.
RecF Pathway
Bacteria appear to use the RecF pathway of homologous recombination to repair single-strand gaps in DNA. When the RecBCD pathway is inactivated by mutations and additional mutations inactivate the SbcCD and ExoI nucleases, the RecF pathway can also repair DNA double-strand breaks. In the RecF pathway the RecQ helicase unwinds the DNA and the RecJ nuclease degrades the strand with a 5’ end, leaving the strand with the 3’ end intact. RecA protein binds to this strand and is either aided by the RecF, RecO, and RecR proteins or stabilized by them. The RecA nucleoprotein filament then searches for a homologous DNA and exchanges places with the identical or nearly identical strand in the homologous DNA.
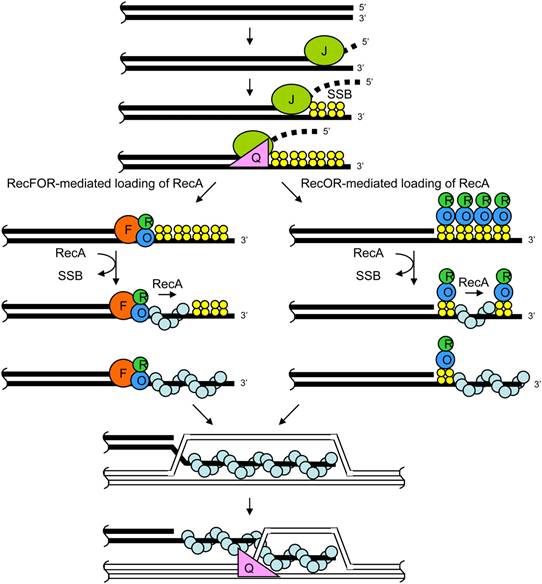
Although the proteins and specific mechanisms involved in their initial phases differ, the two pathways are similar in that they both require single-stranded DNA with a 3’ end and the RecA protein for strand invasion. The pathways are also similar in their phases of branch migration, in which the Holliday junction slides in one direction, and resolution, in which the Holliday junctions are cleaved apart by enzymes. The alternative, non-reciprocal type of resolution may also occur by either pathway.
The exonuclease activity of RecJ resects duplex DNA by degrading in the 5′ → 3′ direction, stimulated by RecQ helicase and SSB. The resultant ssDNA tail is bound by SSB. The RecFOR complex binds at the ssDNA–dsDNA junction on the resected DNA, and loads RecA onto the ssDNA. The RecOR complex can also mediate exchange of RecA for SSB bound to ssDNA at sites away from the junction. The RecA-ssDNA nucleoprotein filament invades homologous recipient duplex DNA. RecJ can contribute to stabilization of the D-loop by degrading the displaced ssDNA after nicking whereas RecQ helicase may disrupt recombination intermediates by unwinding from 3′ → 5′ direction. At this point, the joint molecule can be converted to a Holliday junction and resolved, or disrupted after DNA synthesis to anneal with the second end of a dsDNA break.
Branch
Migration and Resolution (RuvABC Pathway)
Immediately after strand invasion, the Holliday junction moves along the linked DNA during the branch migration process. It is in this movement of the Holliday junction that base pairs between the two homologous DNA duplexes are exchanged. To catalyze branch migration, the RuvA protein first recognizes and binds to the Holliday junction and recruits the RuvB protein to form the RuvAB complex. Two sets of the RuvB protein, which each form a ring-shaped ATPase, are loaded onto opposite sides of the Holliday junction, where they act as twin pumps that provide the force for branch migration. Between those two rings of RuvB, two sets of the RuvA protein assemble in the center of the Holliday junction such that the DNA at the junction is sandwiched between each set of RuvA. The strands of both DNA duplexes—the "donor" and the "recipient" duplexes—are unwound on the surface of RuvA as they are guided by the protein from one duplex to the other.
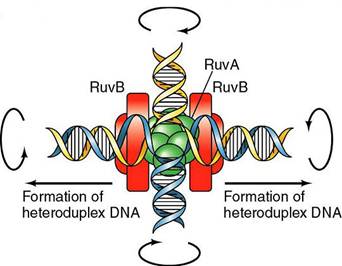
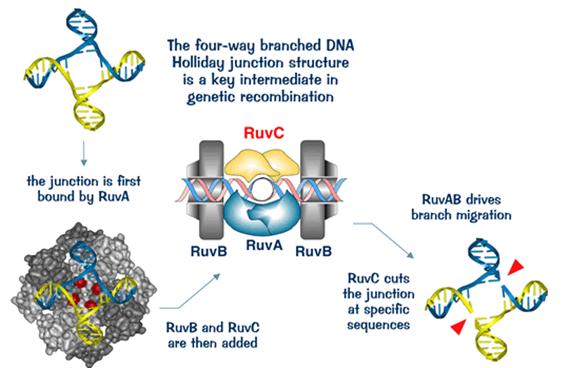
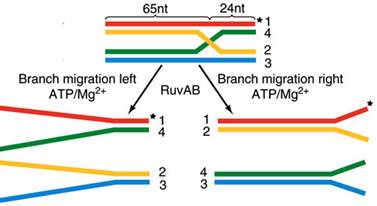
In the resolution phase of recombination, any Holliday junctions formed by the strand invasion process are cut, thereby restoring two separate DNA molecules. This cleavage is done by RuvAB complex interacting with RuvC, which together form the RuvABC complex. RuvC is an endonuclease that cuts the degenerate sequence 5'-(A/T)TT(G/C)-3'. The sequence is found frequently in DNA, about once every 64 nucleotides. Before cutting, RuvC likely gains access to the Holliday junction by displacing one of the two RuvA tetramers covering the DNA there. Recombination results in either "splice" or "patch" products, depending on how RuvC cleaves the Holliday junction. Splice products are crossover products, in which there is a rearrangement of genetic material around the site of recombination. Patch products, on the other hand, are non-crossover products in which there is no such rearrangement and there is only a "patch" of hybrid DNA in the recombination product.
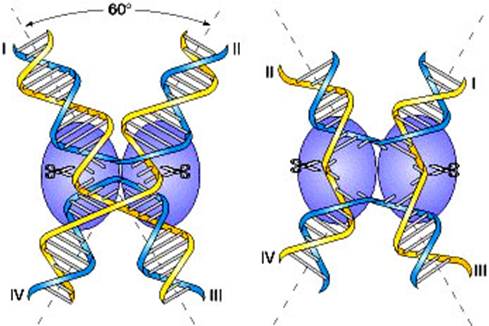
Possible
pathways for processing Holliday junction in E. coli.
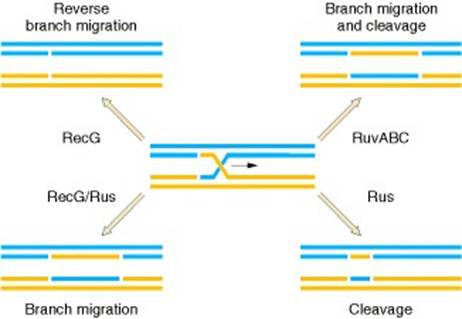
The center of the figure shows a Holliday junction formed by homologous pairing and strand exchange by the RecA protein. In the presence of RecA, branch migration is in the same direction as the RecA strand exchange and is promoted by RuvAB. RuvC cleavage resolves this structure (upper right). Also in the presence of RecA, RecG could drive the Holliday junction backward (upper right). However, if RecA dissociates from the junction, RecG could drive the reaction forward, with resolution coming from the Rus protein (lower left). The lower right shows the results of Rus cleavage alone, after strand exchange and branch migration by RecA.
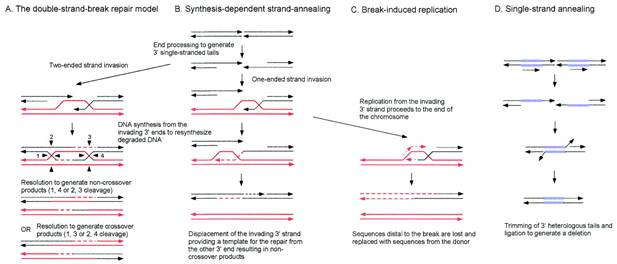
Site
Specific Recombination
Site-specific recombination moves specialized nucleotide sequences, called mobile genetic elements, between nonhomologous sites within a genome. The movement can occur between two different positions in a single chromosome, as well as between two different chromosomes.
Mobile genetic elements range in size from a few hundred to tens of thousands of nucleotide pairs, and they have been identified in virtually all cells that have been examined. Some of these elements are viruses in which site-specific recombination is used to move their genomes into and out of the chromosomes of their host cell. A virus can package its nucleic acid into viral particles that can move from one cell to another through the extracellular environment. Many other mobile elements can move only within a single cell (and its descendents), lacking any intrinsic ability to leave the cell in which they reside.
The relics of site-specific recombination events can constitute a considerable fraction of a genome. The abundant repeated DNA sequences found in many vertebrate chromosomes are mostly derived from mobile genetic elements; in fact, these sequences account for more than 45% of the human genome. Over time, the nucleotide sequences of these elements have been altered by random mutation. As a result, only a few of the many copies of these elements in our DNA are still active and capable of movement.
In addition to moving themselves, all types of mobile genetic elements occasionally move or rearrange neighboring DNA sequences of the host cell genome. These movements can cause deletions of adjacent nucleotide sequences, for example, or can carry these sequences to another site. In this way, site-specific recombination, like general recombination, produces many of the genetic variants upon which evolution depends. The translocation of mobile genetic elements gives rise to spontaneous mutations in a large range of organisms including humans; in some, such as the fruit fly Drosophila, these elements are known to produce most of the mutations observed. Over time, site-specific recombination has thereby been responsible for a large fraction of the important evolutionary changes in genomes.
Mobile
genetic elements
Each of these DNA elements contains a gene
that encodes a transposase, an enzyme that conducts at least some of the
DNA breakage and joining reactions needed for the element to move. Each mobile
element also carries short DNA sequences (indicated in dark) that are
recognized only by the transposase encoded by that element and are necessary
for movement of the element. In addition, two of the three mobile elements
shown carry genes that encode enzymes that inactivate the antibiotics
ampicillin (ampR) and tetracycline (tetR). The transposable element Tn10, shown in the
bottom diagram, is thought to have evolved from the chance landing of two short
mobile elements on either side of a tetracyclin-resistance
gene; the wide use of tetracycline as an antibiotic has aided the spread of
this gene through bacterial populations. The three mobile elements shown are
all examples of DNA-only transposons.
Mechanism for site specific recombinations
Site-specific recombination can proceed via either of two distinct mechanisms, each of which requires specialized recombination enzymes and specific DNA sites.
(1) Transpositional site-specific recombination usually involves breakage reactions at the ends of the mobile DNA segments embedded in chromosomes and the attachment of those ends at one of many different nonhomologous target DNA sites. It does not involve the formation of heteroduplex DNA.
(2) Conservative site-specific recombination involves the production of a very short heteroduplex joint, and it therefore requires a short DNA sequence that is the same on both donor and recipient DNA molecules.
Transpositional site-specific recombination (Transposons)
Transposons, also called transposable elements, are mobile genetic elements that generally have only modest target site selectivity and can thus insert themselves into many different DNA sites. In transposition, a specific enzyme, usually encoded by the transposon and called a transposase, acts on a specific DNA sequence at each end of the transposon—first disconnecting it from the flanking DNA and then inserting it into a new target DNA site. There is no requirement for homology between the ends of the element and the insertion site. Most transposons move only very rarely (once in 105 cell generations for many elements in bacteria), and for this reason it is often difficult to distinguish them from nonmobile parts of the chromosome.
On the basis of their structure and transposition mechanisms, transposons can be grouped into three large classes, each of which is discussed in detail in subsequent sections. Those in the first two of these classes use virtually identical DNA breakage and DNA joining reactions to translocate. However, for the DNA-only transposons, the mobile element exists as DNA throughout its life cycle: the translocating DNA segment is directly cut out of the donor DNA and joined to the target site by a transposase. In contrast, retroviral-like retrotransposons move by a less direct mechanism. An RNA polymerase first transcribes the DNA sequence of the mobile element into RNA. The enzyme reverse transcriptase then transcribes this RNA molecule back into DNA using the RNA as a template, and it is this DNA copy that is finally inserted into a new site in the genome. For historical reasons, the transposase-like enzyme that catalyzes this insertion reaction is called an integrase rather than a transposase. The third type of transposon also moves by making a DNA copy of an RNA molecule that is transcribed from it. However, the mechanism involved for these nonretroviral retrotransposons is distinct from that just described in that the RNA molecule is directly involved in the transposition reaction.
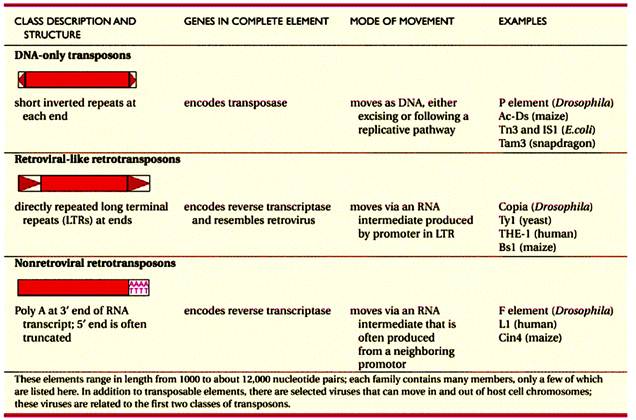
DNA
only Transposons
Cut
and Past transposition
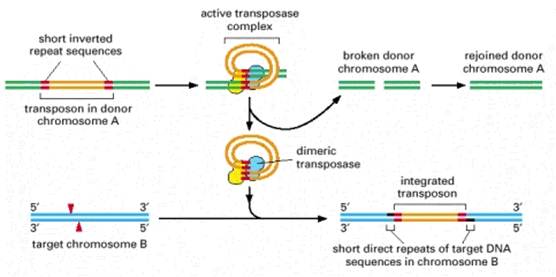
DNA-only transposons can be recognized in chromosomes by the “inverted repeat DNA sequences” at their ends. Experiments show that these sequences, which can be as short as 20 nucleotides, are all that is necessary for the DNA between them to be transposed by the particular transposase enzyme associated with the element. The cut-and-paste movement of a DNA-only transposable element from one chromosomal site to another begins when the transposase brings the two inverted DNA sequences together, forming a DNA loop. Insertion into the target chromosome, catalyzed by the transposase, occurs at a random site through the creation of staggered breaks in the target chromosome. As a result, the insertion site is marked by a short direct repeat of the target DNA sequence, as shown. Although the break in the donor chromosome is resealed, the breakage-and-repair process often alters the DNA sequence, causing a mutation at the original site of the excised transposable element.
Replicative
transposition
Some DNA-only transposons move using a variation of the cut-and-paste mechanism called replicative transposition. In this case, the transposon DNA is replicated and a copy is inserted at a new chromosomal site, leaving the original chromosome intact. In the course of replicative transposition, the DNA sequence of the transposon is copied by DNA replication. The end products are a DNA molecule that is identical to the original donor and a target DNA molecule that has a transposon inserted into it.
Retroviral
like retrotransposons
Certain viruses are considered mobile genetic elements because they use transposition mechanisms to integrate their genomes into that of their host cell. The bacteriophage Mu not only uses DNA-based transposition to integrate its genome into its host cell chromosome, it also uses the transposition process to initiate its viral DNA replication. The Mu transposase was the first to be purified in active form and characterized; it recognizes the sites of recombination at each end of the viral DNA by binding specifically to this DNA.
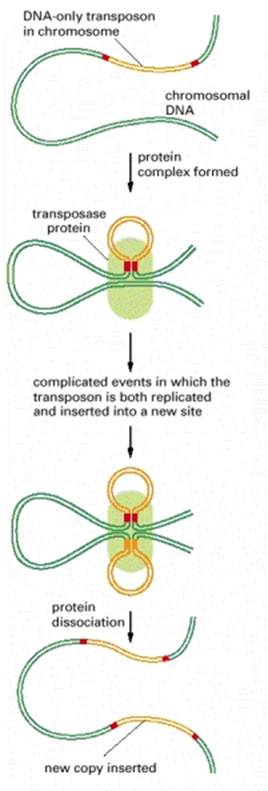
Transposition also has a key role in the life cycle of many other viruses. Most notable are the retroviruses, which include the AIDS virus, called HIV, that infects human cells. Outside the cell, a retrovirus exists as a single-stranded RNA genome packed into a protein capsid along with a virus-encoded reverse transcriptase enzyme. During the infection process, the viral RNA enters a cell and is converted to a double-stranded DNA molecule by the action of this crucial enzyme, which is able to polymerize DNA on either an RNA or a DNA template. The term retrovirus refers to the fact that these viruses reverse the usual flow of genetic information, which is from DNA to RNA. Specific DNA sequences near the two ends of the double-stranded DNA product produced by reverse transcriptase are then held together by a virus-encoded integrase enzyme. This integrase creates activated 3′-OH viral DNA ends that can directly attack a target DNA molecule through a mechanism very similar to that used by the cut-and-paste DNA-only transposons.
Outline of the strand-breaking and strand-rejoining events that lead to integration of the linear double-stranded DNA of a retrovirus (such as HIV) or a retroviral-like retrotransposon (such as Ty1) into the host cell chromosome (blue). In an initial step, the integrase enzyme forms a DNA loop and cuts one strand at each end of the viral DNA sequence, exposing a protruding 3′-OH group. Each of these 3′-OH ends then directly attacks a phosphodiester bond on opposite strands of a randomly selected site on a target chromosome. This inserts the viral DNA sequence into the target chromosome, leaving short gaps on each side that are filled in by DNA repair processes. Because of the gap filling, this type of mechanism (like that of cut-and-paste transposons) leaves short repeats of target DNA sequence on each side of the integrated DNA segment; these are 3–12 nucleotides long, depending on the integrase enzyme.
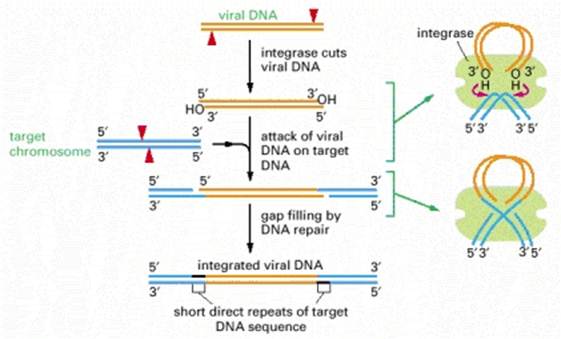
Non-retroviral
retrotransposons
A significant fraction of many vertebrate chromosomes is made up of repeated DNA sequences. In human chromosomes, these repeats are mostly mutated and truncated versions of a retrotransposon called an L1 element (sometimes referred to as a LINE or long interspersed nuclear element). Although most copies of the L1 element are immobile, a few retain the ability to move. Translocations of the element have been identified, some of which result in human disease; for example, a particular type of hemophilia results from an L1 insertion into the gene encoding a blood clotting factor, Factor VIII. Related mobile elements are found in other mammals and insects, as well as in yeast mitochondria. These nonretroviral retrotransposons move via a distinct mechanism that requires a complex of an endonuclease and a reverse transcriptase.
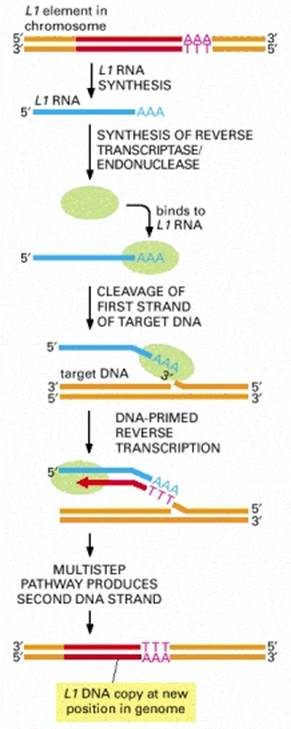
Transposition by the L1 element begins when an endonuclease attached to the L1 reverse transcriptase and the L1 RNA makes a nick in the target DNA at the point at which insertion will occur. This cleavage releases a 3′-OH DNA end in the target DNA, which is then used as a primer for the reverse transcription step shown. This generates a single-stranded DNA copy of the element that is directly linked to the target DNA. In subsequent reactions, not yet understood in detail, further processing of the single-stranded DNA copy results in the generation of a new double-stranded DNA copy of the L1 element that is inserted at the site where the initial nick was made.
Conservative site-specific
recombination
In this pathway, breakage and joining
occur at two special sites, one on each participating DNA molecule. Depending
on the orientation of the two recombination
sites, DNA integration, DNA excision, or DNA inversion can occur. Site-specific
recombination enzymes that break and rejoin
two DNA double helices at specific sequences on each DNA molecule often do so
in a reversible way: the same enzyme system that joins two DNA molecules can
take them apart again, precisely restoring the sequence of the two original DNA
molecules. This type of recombination is
therefore called “conservative” site-specific
recombination.
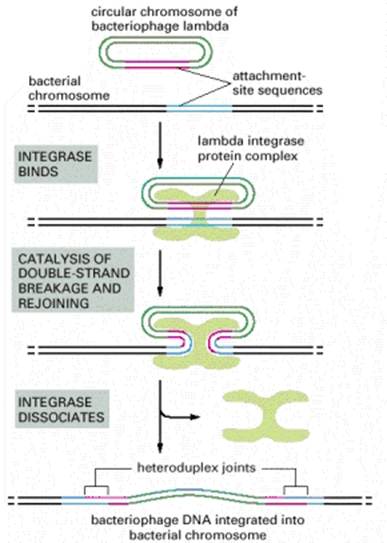
In this example of site-specific recombination, the lambda integrase enzyme binds to a specific “attachment site” DNA sequence on each chromosome, where it makes cuts that bracket a short homologous DNA sequence. The integrase then switches the partner strands and reseals them to form a heteroduplex joint that is seven nucleotide pairs long. A total of four strand-breaking and strand-joining reactions is required; for each of them, the energy of the cleaved phosphodiester bond is stored in a transient covalent linkage between the DNA and the enzyme, so that DNA strand resealing occurs without a requirement for ATP or DNA ligase.
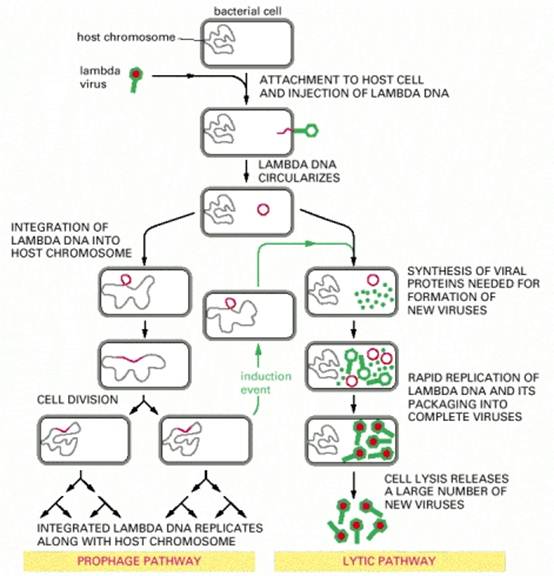
The same type of site-specific recombination mechanism can also be used in reverse to promote the excision of a mobile DNA segment that is bounded by special recombination sites present as direct repeats. In bacteriophage lambda, excision enables it to exit from its integration site in the E. coli chromosome in response to specific signals and multiply rapidly within the bacterial cell. Excision is catalyzed by a complex of integrase enzyme and host factors with a second bacteriophage protein, excisionase, which is produced by the virus only when its host cell is stressed—in which case, it is in the bacteriophage's interest to abandon the host cell and multiply again as a virus particle.
Conservative site-specific
recombination and
Gene turn on or off
When the special sites recognized by a conservative site-specific recombination enzyme are inverted in their orientation, the DNA sequence between them is inverted rather than excised. Such inversion of a DNA sequence is used by many bacteria to control the gene expression of particular genes—for example, by assembling active genes from separated coding segments. This type of gene control has the advantage of being directly inheritable, since the new DNA arrangement is transferred to daughter chromosomes automatically when a cell divides.
This technique requires the insertion of two specially engineered DNA molecules into the animal's germ line. (A) The DNA molecule shown has been engineered with specific recognition sites so that the gene of interest is transcribed only after a site-specific recombination enzyme that uses these sites is induced. As shown on the right, this induction removes a marker gene and brings the promoter DNA (orange) adjacent to the gene of interest. The recombination enzyme is inducible, because it is encoded by a second DNA molecule (not shown) that has been engineered to ensure that the enzyme is made only when the animal is treated with a special small molecule or its temperature is raised. (B) Transient induction of the recombination enzyme causes a brief burst of synthesis of that enzyme, which in turn causes a DNA rearrangement in an occasional cell. For this cell and all its progeny, the marker gene is inactivated and the gene of interest is simultaneously activated (as shown in A). Those clones of cells in the developing animal that express the gene of interest can be identified by their loss of the marker protein. This technique is widely used in mice and Drosophila, because it allows one to study the effect of expressing any gene of interest in a group of cells in an intact animal. In one version of the technique, the Cre recombination enzyme of bacteriophage P1 is employed along with its loxP recognition sites.
**********